

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Adrenalectomy is an integral procedure of both general and urological surgeons. The finding of a large, progressively growing adrenal mass that is enhanced on CT always arouses concern for an underlying malignant tumor and leads to the scheduling of prompt surgical resection.
In rare cases, however, malignant-appearing masses turn out to be benign, one of which is an adrenal ganglioneuroma (AGN). Ganglioneuromas are large neoplasms originating within the neural crest and consisting of ganglion cells, Schwann cells, and nerve fibers [1]. The adrenal gland is the most involved site [2].
The incidence of AGN is reported to vary between 0.3% to 4% of all adrenal masses [13], or 1 per million in the general population [2]. There are many case reports of AGN in the English literature. One study found 41 AGN cases reported between 1961 and 2009 [4], another counted 230 cases between 1978 and 2008 [5], while a third study found 150 cases between 2000 and 2016 [1].
As of 2021, there are only 5 case series in the English literature reporting over 20 cases and originating in China, South Korea, and the United States [36789]; another 3 originating in Italy and China and report 10–17 cases each [11011] and 4 case series of 6–9 cases each from Canada [4], Italy [2], Greece [5], and Belgium [12]. The largest studies from the Far East and the United States are single-center experiences and relate to 29—45 cases recorded between 1988 and 2019 [36789].
In the present study, we sought to retrospectively review the collective experience with AGN pathologically confirmed following adrenalectomy in 4 medium- to high-volume medical centers in Israel. To the best of our knowledge, this is the largest study on this topic coming from the Middle East region.
METHODS
Four medical centers participated in the present study. Following approval of each medical center's institutional review board, all adrenalectomy cases registered between 2006 and 2020 were retrospectively reviewed. The recorded data included patients' age, sex, tumor side, relevant family history, radiological and laboratory findings, associated clinical symptoms, date of surgery and surgical technique, maximal diameter of the tumor and other tumor components according to pathological findings, tumor recurrence, and follow-up duration. Data acquired from all medical centers were integrated and interpreted. The results are given in average (range) unless otherwise specified.
This study has been approved by the Institutional Review Board of Chaim Sheba Medical Center (No. SMC-18-5818) with a waiver for informed consent.
RESULTS
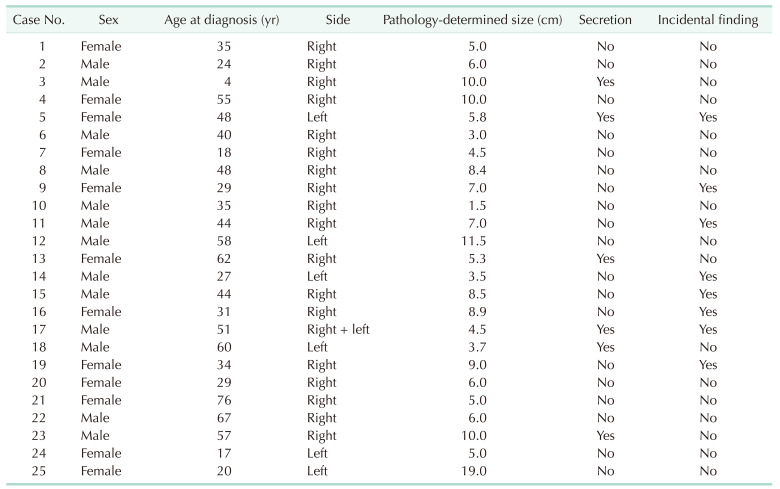
A total of 875 adrenalectomy cases were reviewed between 2006 and 2020, among which the final pathology was AGN in 25 patients (2.9%). The demographic data of those patients are detailed in Table 1. Their average age was 40.5 years (range, 4–76 years), and 13 (52.0%) were males.
The AGNs of 8 patients (32%) who were entirely asymptomatic were found incidentally. The clinical symptoms of the remaining 17 patients were as follows; abdominal pain/discomfort (11 patients, 44.0%), back pain (3 patients, 12.0%), weight loss (3 patients, 12.0%), and hot flashes and hypertension (1 patient each, 4.0%). Eighteen patients (72.0%) had right-side lesions, and 1 patient had bilateral AGNs. One patient had a family history of neurofibromatosis (NF) 1, and 1 patient had a succinate dehydrogenase (SDHD) gene mutation.
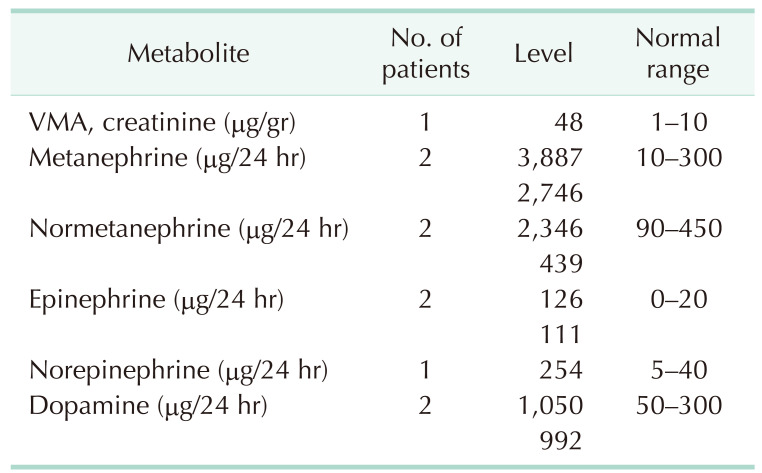
Only 6 cases (24.0%) had pathological metabolite hypersecretion detected in 24-hour urine collection, specifically, vanillylmandelic acid (1 observation) and catecholamines (CA; epinephrine, norepinephrine, metanephrine, normetanephrine, and dopamine) (9 observations in total). The specific levels, as well as normal ranges, are detailed in Table 2.
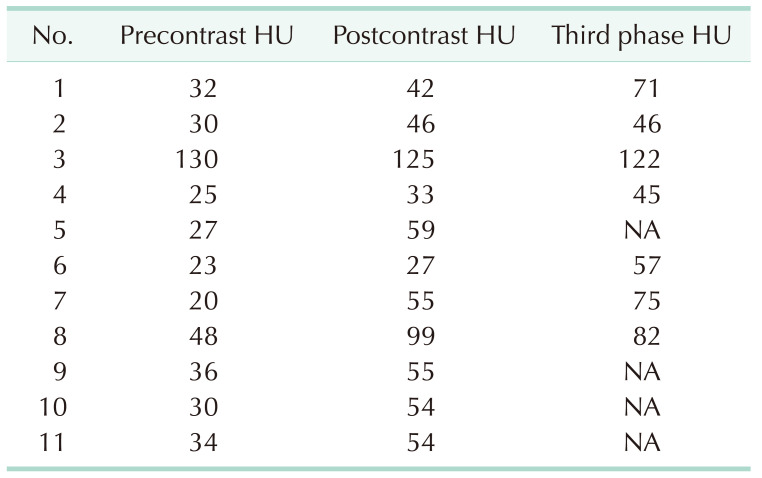
The radiological diagnosis was based on a CT scan in all 25 cases. The estimated average maximal tumor diameter on CT was 6.61 cm (range, 1.5–20 cm). Pre- and postcontrast Hounsfield units (HU) as well as late-phase HU were available for 11 patients, and the postcontrast HU values were higher than the precontrast ones in 10 of them (Table 3). The average pre- and postcontrast HU values were 35.2 and 59.0, respectively, and the average late-phase HU value was 71.1. Tumor calcification was detected in 4 cases (16.0%).
Twenty-two patients (88.0%) underwent minimally invasive surgery for removal of the adrenal mass (21 by laparoscopy and 1 by robot-assisted surgery), while the remaining 3 had open surgery. The average tumor diameter in the final pathology report was 7 cm (range, 1.5–19.0 cm). Isolated AGN was diagnosed in 21 cases (84.0%), and the additional components reported for the remaining 4 cases included pheochromocytoma (PC) in 2, ganglioneuroblastoma (GNB) in 1, and neurofibroma in 1.
Positive surgical margins were detected in 3 cases (12.0%). Notably, a coexisting high secretion of CA was detected in 2 of the 3 cases in which a PC component was present, and coexisting hypertension was observed in the 3rd case.
The average follow-up duration for the entire group was 16.8 months (range, 1–136 months). All 25 study patients were free from local recurrence and metastatic progression. Data acquired from the national population registry revealed that they all were alive at study closure.
DISCUSSION
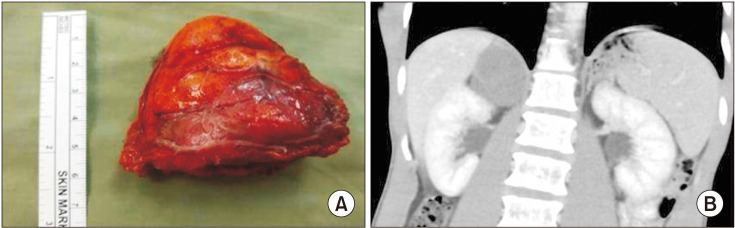
The international neuroblastoma (NB) pathology classification divides neuroblastic tumors into 4 subcategories based on their morphology, clinical features, and behavior; NB, nodular GNB, intermixed GNB, and ganglioneuroma [13]. Ganglioneuroma is a rare, differentiated neuroblastic tumor of the adrenal gland originating in the neural crest cells [14]. It is found within the adrenal medulla in 20%–0% of the cases [4]. They are macroscopically described as lobulated and gray-white [12], although they also appeared encapsulated and well-demarcated in this and other studies [811] (Fig. 1).
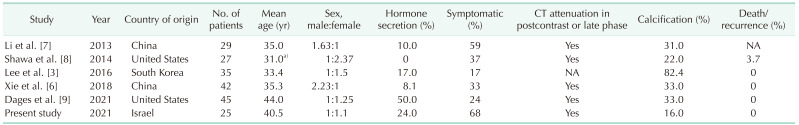
Table 4 summarizes the findings of the present study in comparison to those found in previous large case series (i.e., ≥20 cases). As demonstrated in the present and previous case series [1367891011], the peak incidence of AGN is the 4th–5th decade of life [1314].
A male-to-female ratio of 2:1 has been reported for adrenomedullary tumors [15]; however, an equal ratio for both sexes has been found in the present study as well as in other studies [914]. AGN has been reported to accompany hereditary familial syndromes, such as NF1 and NF2 [1214], and 1 study also reported a single AGN case with an associated multiple endocrine neoplasia 2 syndrome [8].
We had 1 case associated with NF1 and another case with SDHD gene mutation. Most AGNs are hormonally silent [314]; however, some of them may be associated with hormonal secretion. Glucocorticosteroid (i.e., cortisol), mineralocorticoid (i.e., aldosterone), and androgen (i.e., testosterone and its precursors) secretion have been reported in numerous cases [1467].
Secretion of CA and their metabolites have been observed in our study as well as in others [234567]. In the present cohort, pathological CA metabolite secretion was observed in 24% of our cases. This may be partly explained by the origination of AGN in the adrenal cortex rather than the adrenal medulla. Another explanation could derive from the PC component known to be associated with some of the AGN originating within the medulla [415]. In our study, a PC component was present in 3 cases of which 2 demonstrated relatively high urinary CA secretion.
Notwithstanding, the extent of hormone secretion does not always suffice to produce clinical symptoms. In our study, hypertension was present in 1 of the 3 cases producing CA, similar to the findings of other studies [3467].
Cortisol secretion was not associated with clinical symptoms in 1 study [1] but reported to have led to menstrual disorders in another study [6]. Rondeau et al. [4] described virilization in 1 case in which increased testosterone secretion was detected, and Li et al. [7] described central obesity due to urinary 17-hydroxysteroid hypersecretion.
Interestingly, many reports describe AGN as an asymptomatic, incidental finding. Asymptomatic cases reportedly vary between 71.4% and 100% [12345612], although in the same breath symptoms attributable to AGN, i.e., most commonly, abdominal pain, back pain, and abdominal discomfort [12361112]; and less commonly fatigue and malaise [711] have also been mentioned. In our study, similar to the findings of Shawa et al. [8], abdominal and back symptoms were relatively common (56%), with a mass effect having been suggested as a possible mechanism [38].
A CT scan is the most common imaging tool implemented to characterize an adrenal mass. An AGN as demonstrated on a CT scan is homogenous and of low attenuation in the precontrast phase, mild attenuation in the postcontrast phase, and sometimes either homogenous or heterogeneous in appearance in the ate phase [311]. This pattern was repeatedly observed by us as well as by others [4568912]. Calcification is another indication for AGN seen on CT; however, its prevalence widely varies between studies from 11.1% [2] to as high as 82.4% [3]. It should be kept in mind however, that calcifications, heterogeneity, and postcontrast attenuation are seen in malignant adrenal tumors as well [16].
MRI has been suggested as an alternative diagnostic tool for AGN. Low signal intensity on T1 and high signal intensity on T2-weighted images are expected [27], but the malignant tumor cannot be excluded, and the final diagnosis is most definitive by surgical histology.
AGN develops in childhood but it is not recognized before early adulthood, and it is rarely diagnosed after the age of 60 years [2]. These findings are in line with the single patient under 10 years of age (i.e., a 4-year-old) as well as the 3 patients who were older than 60 years of age at diagnosis in our cohort.
The tumor rarely metastasizes or recurs [14]. No tumor recurrence or death due to AGN was observed in the present study or in any other studies in the literature [123456891011], although 1 mortality in association with AGN was reported due to CA hypersecretion [17]. While the overall prognosis is excellent, surgical excision should remain the standard of care due to the inability of radiology to discriminate between AGN from a malignant tumor.
The present study summarizes the experience of 4 medium-to high-volume medical centers throughout a period of 14 years. To the best of our knowledge, this is the first case series on this topic coming from the Middle East region and is among the largest AGN case series published in English literature. Its retrospective nature and relatively low case numbers are its main disadvantages. Another weak point is that there was no standardization of the CT scans, or of the laboratory and histopathology results among the participating institutions, which could potentially affect the interpretation of the main findings.
In summary, AGN is a rare, slow-growing, large benign tumor with peak occurrence during the 4th–5th decade of life. There is no gender predominance. The tumor may be hormone-secreting, however not necessarily to the level that would create associated symptoms. The most common symptom is abdominal/back pain which is attributed to a mass effect. Given its radiologic characteristics, which cannot be discriminated from a malignant adrenal tumor, the final diagnosis should be made by surgical resection, preferably laparoscopic if possible. The overall prognosis of AGN is excellent.
 XML Download
XML Download