

 PDF
PDF Citation
Citation Print
Print



Introduction
Thin glomerular basement membrane (GBM) nephropathy is characterized by thinning of the GBM and is associated with structural abnormality of α-chains of type IV collagen [1]. The disease is diagnosed based on histologic examination of kidney biopsy, and thinning of the GBM is seen on an electron microscope. Clinical manifestations are hematuria, mild proteinuria with normal kidney function and the disease itself has benign clinical course [2]. Among the two-thirds of patients with thin GBM nephropathy, hematuria is observed in other family members showing an autosomal dominant inheritance [3]. Forty to fifty percent of the patients are accompanied by heterozygous COL4A3 or COL4A4 mutations with incomplete penetrance in 5–10% [4]. However, it has been observed that a few patients with heterozygous COL4A3/COL4A4 mutation, showed a progressive clinical course that may progress to end-stage renal disease [5].
Alport syndrome (AS) is caused by mutations in the COL4A3/A4/A5 gene, and most cases harbor mutations in the COL4A5 gene located in the X chromosome. AS is associated with progressive kidney failure, hearing loss and eye abnormalities. There are several well-established studies regarding eye abnormalities in AS [6]; however, in autosomal dominant AS (ADAS), less than 3% of the patients have been reported to have ocular abnormalities [2]. Therefore, the involvement of ocular abnormalities may be helpful in the differential diagnosis of these two diseases.
Generalization of whole exome sequencing (WES) may lead to a lot of confusion and difficulties in making differential diagnosis due to various clinical phenotypes, pathological findings, and prognosis of patients with heterozygous COL4A3/COL4A4 mutations. Therefore, a new classification of diseases resulting from COL4A3/COL4A4 /COL4A5 mutations also known as AS has been introduced recently. According to this classification, thin GBM disease has been no more used and ADAS has been replaced the misnomer [7,8]. However, clinically most of the patients with heterozygous COL4A3 or COL4A4 mutations show hematuria with GBM thinning, and there are no main features of AS, such as progressive kidney failure, deafness, and ocular abnormalities. The classification itself may give rise to anxiety in this disease group and people with the disease oppose to be named as ADAS [9]. Therefore, careful consideration and appropriateness of classification are required for the disease classification in patients with COL4A3/COL4A4 mutations in near future.
Morning Glory syndrome (MGS) is a congenital optic disc anomaly first reported in the 1970s [10]. MGS commonly have isolated ocular abnormalities, however, systemic associations such as Aicardi syndrome have also been reported [11,12]. MGS associated with heterozygous COL4A3/A4 mutations have not been reported in the literature.
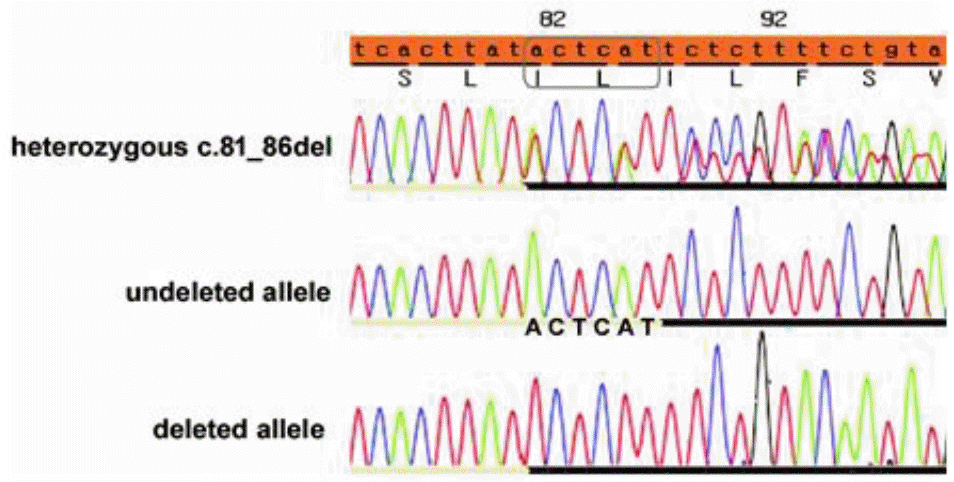
Here, we report the first case of ADAS with a heterozygous COL4A4 mutation [c.81_86del (p.Ile29_Leu30del)] associated with MGS.
Case report
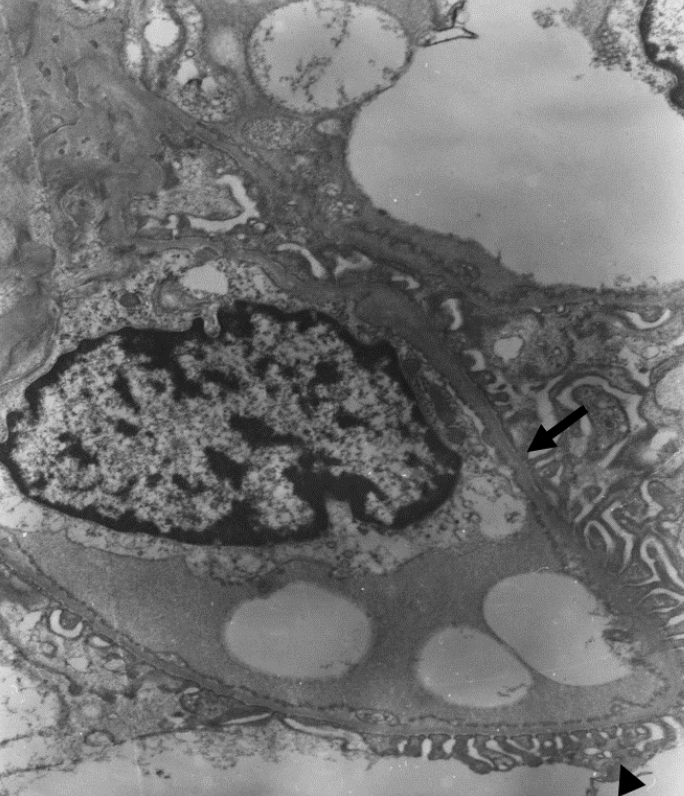
A 6-year-old male patient visited the nephrology clinic because of incidental findings of microscopic hematuria in school medical check-up. There were no accompanying clinical symptoms beside hematuria (red blood cells 20–30/high power field) with normal kidney function [Creatinine (Cr); 0.43 mg/dL, estimated glomerular filtration rate (eGFR); 140 ml/min/1.73m2]. After his first visit, he had annual follow-ups, and at the age of 12 years, he was diagnosed with ADAS by kidney biopsy (Fig. 1). Meanwhile, because of progressive deterioration of visual acuity, he visited the ophthalmology clinic and was diagnosed with MGS on the left eye; eventually, there was almost loss of vision in his left eye. His urine examination revealed hematuria with mild proteinuria but normal kidney function. No hypertension was also noted. He and his family members underwent WES and heterozygous COL4A4 mutations [NM_000092.4:c.81_86del (p.Ile29_Leu30del)] were found in the patient and his mother (Fig. 2). There was no family history of chronic kidney disease in both of his parents but, his mother and sister showed microscopic hematuria on urine examination. Otherwise, they had normal kidney function, and no other abnormalities were found during eye examinations. As COL4A4 mutation was the only result of WES in this patient, there are no possibilities for other genetic mutations took part in this patient’s nephropathy and eye lesion.
1. Molecular genetic testing
We extracted DNA samples from the oral mucosal epithelium of the patient and established a library using xGen Exome Research Panel v2 (Integrated DNA Technologies, Coralville, Iowa, USA). WES (~22000) was performed using Novaseq 6000 (Illumina, San Diego, CA, USA). Sequencing was performed using a self-manufactured pipeline by combining precedent bioinformatics tool, and this was evaluated according to the American College of Medical Genetics and Genomics/the Association for Molecular Pathology guidelines. To reconfirm the mutation in the COL4A4 gene detected in the exome test, DNA extracted from the peripheral blood of the patient and parent was used to perform sequencing of the relevant site. BigDye Terminator v3.1 (Applied Biosystems, Foster City, California, USA) dye and ABI3500 (Applied Biosystems) were used.
2. Ophthalmologic examination
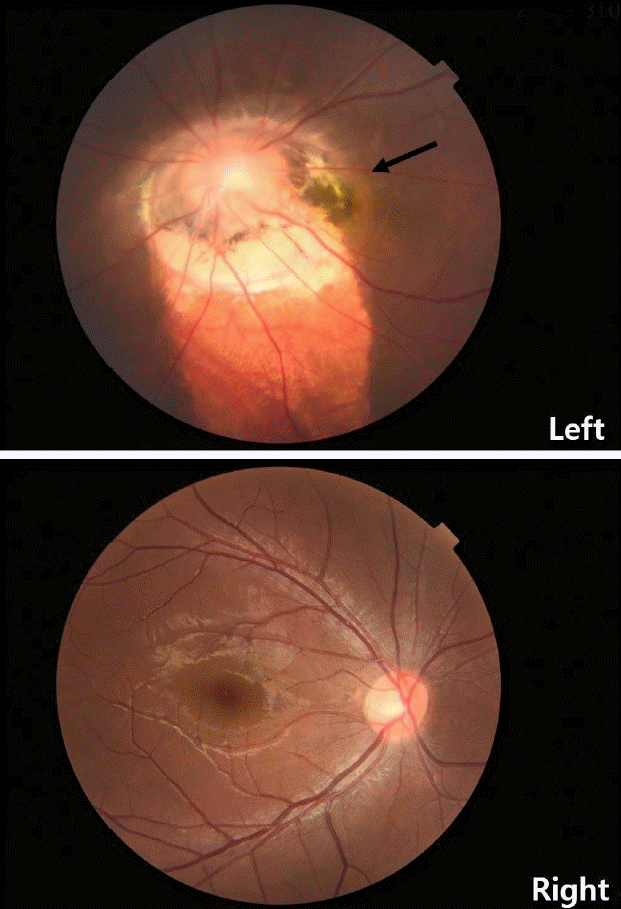
In the ophthalmologic examination, the right eye showed -2.5 sph -0.75 cyl A180 and the left eye showed -5.25 Sph -0.5 cyl A150. In the refractive examination, best corrected visual acuity was 1.5 in the right eye, and 0.05 in the left eye. Slit-lamp examination showed no abnormality; however, fundoscopic examination revealed enlarged optic papillae and radially stretched papillary vessels (Fig. 3). There were no signs of retinal detachment on the retinal tomography.
Discussion
Patients with heterozygous COL4A3/A4 mutations have a broad clinical spectrum ranging from familial isolated microscopic hematuria to end-stage renal disease and are diagnosed as thin GBM nephropathy, ADAS, and familial focal segmental glomerulosclerosis [13]. However, diagnostic difficulties still exist, and clinical manifestations and outcomes have not yet been established. In particular, when heterozygous COL4A3/A4 mutations are recognized during childhood and adolescence, the appropriateness of the initial diagnosis may need to be re-evaluated depending on the clinical course of the patient as they age.
In the early stage of AS, some patients show only GBM thinning same as in patients with thin GBM nephropathy. And when a kidney biopsy is performed at a younger age, it is difficult to diagnose GBM thinning based on age-appropriate diagnostic criteria. Clinically, isolated microscopic hematuria can be seen in the early stage of AS, similar to ADAS. In ADAS, mild proteinuria is present in some patients; however, kidney function is well-preserved and progression to end-stage renal disease is rare. Extra-kidney manifestations, including ocular features, are rarely reported; therefore, the presence of these symptoms is sometimes helpful in making a differential diagnosis of AS from ADAS.
Owing to the generalization of molecular genetic testing, recent research results have shown that some groups of patients with heterozygous COL4A3/A4 mutations with benign familial hematuric disease may have a more serious progressive course than previously known benign course. In a study, of the patients with heterozygous COL4A3/A4 mutations (777 patients from 258 families), 94.8% of the patients had hematuria, and 29% of the patients had impaired kidney function. Moreover, 52.3% of the patients with chronic kidney disease, eventually progressed to end-stage renal disease and ocular findings were observed in only 3% of patients [2]. In AS, many studies have reported accompanying ocular features, which are well established [6]. However, there are few studies related to ocular findings in ADAS, and most of the patients who underwent ophthalmic examinations had no notable abnormalities [2,3]. There is no description of ocular phenotype in a case with same genetic mutation as in this case [14].
MGS was named by Kindler in 1970 because of the shape resemblance of abnormal optic nerve papilla to the morning glory flower and were reported by Krause, Steinkuller, and Beyer [10]. It is characterized by characteristic fundal findings with severe visual impairment. The etiology of MGS is thought to be a congenital mesodermal abnormality or an undesired transformation of the optic nerve papilla coloboma. MGS is characterized by a funnel-shaped depression in the central part. The optic nerve disc is expanded and surrounded by a slightly elevated periphery because of chorioretinal pigment changes. Retinal blood vessels extend radially around the optic disc.
This patient was diagnosed with ADAS by the WES and kidney biopsy. To date, mild proteinuria was observed in addition to hematuria; both blood pressure and renal function were well preserved. In the family history, the patient’s mother and younger sister also had hematuria with normal kidney function.
It is more reasonable to consider MGS seen in this patient as a coincidental finding of ADAS. However, the possibility that heterozygous COL4A3/A4 mutations may have caused MGS cannot be completely excluded. In particular, ADAS with heterozygous COL4A3/A4 mutations in the pediatric population necessitates careful observation over a long period of time and evaluation of extra-kidney manifestations must be required.
 XML Download
XML Download