

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
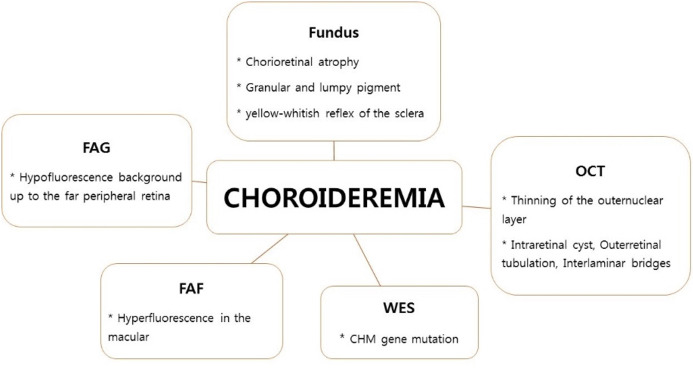
Choroideremia, first described by Mauthner in 1982, refers to the absence (-eremia) of choroid. Choroideremia is an X-linked recessive disease usually characterized by night blindness in male pre-teens.123 The reported global incidence of choroideremia ranges from 1:50,000 to 1:100,000, but there have been no reports on the prevalence of choroideremia in Korea.2345 Choroideremia is caused by mutations in the CHM gene, which maps to Xq21.1-q21.3. This gene consists of 15 exons and encodes the 653-amino acid intracellular protein Rab escort protein-1 (REP-1), which is involved in intracellular vesicle trafficking and recycling.678 Known mutations in CHM include deletions, translocations, and point mutations (nonsense, frameshift, and splice site).
Patients with choroideremia show preservation of visual acuity and the central visual field (VF) for a relatively long time, but progressive loss of peripheral field and visual impairment occur due to degeneration of the retinal pigment epithelium (RPE) and photoreceptor layer. The disease is characterized by fundus findings, VF abnormalities, electroretinogram (ERG) findings, and family history, but is difficult to diagnose because it shows a pattern similar to other X-linked genetic diseases. Therefore, choroideremia is often misdiagnosed as X-linked retinitis pigmentosa (RP), gyrate atrophy, or Kearns–Sayre syndrome.289101112 In previous studies, it was reported that 6% of patients diagnosed with RP had choroideremia.1314
In this study, we performed whole-exome sequencing (WES) of patients initially diagnosed with RP, and their families, and report the clinical characteristics of five patients diagnosed with choroideremia, along with the results of genetic analyses.
METHODS
Subjects
We examined 94 patients initially diagnosed with RP at other hospitals, who visited the Department of Ophthalmology of Soonchunhyang University Bucheon Hospital between November 2017 and February 2018, along with 64 of their family members. Patients affected by any other ocular disorders were excluded from the study. A total of 158 subjects underwent WES to identify the causative gene.
All patients underwent comprehensive ophthalmic evaluation, including best-corrected visual acuity, slit lamp examination, fundus photography, fundus autofluorescence (FAF), fluorescein angiography (FAG), VF, ERG, and optical coherence tomography (OCT). Fundus photography, FAF, and FAG were performed using an Optos P200DTx (Optos PLC, Dunfermline, UK). VF was determined using a Humphrey Field Analyzer 750i (Zeiss Humphrey Systems, Dublin, CA, USA). ERG was measured using VERIS software (Electro-Diagnostic Imaging, Inc., Redwood City, CA, USA). OCT was performed using DRI OCT-1 Atlantis (Topcon Medical Systems Inc., Paramus, NJ, USA).
WES
DNA extraction, libraries construction and sequencing
The genomic DNA was extracted from blood samples following the manufacture’s standard procedure (GeneAll, Seoul, Korea). The integration of DNA was assessed using gel electrophoresis and qubit 2.0 fluorometer (Thermo Fisher Scientific, Waltham, MA, USA) for the DNA degradation as well as contamination and DNA concetration, respectively. A total of 1 μg of high-quality genomic DNA per sample was used in the Agilent SureSelect Human All Exon v5 kit caputure process. Randomly fragmented DNA was end-repaired, extend within an ‘A’ nucleotide at the 3’end, ligated with the indexing-specific paired-end adapter and amplified according to the manufacture’s protocol (Agilent Technologies, Santa Clara, CA, USA). The qualified exome-captured libraries were sequenced aiming throughput at least 100× coverage using HiSeq 4000 according to the manufacturer’s protocol.
Mapping, filtering, calling joint variants
Before calling variants to avoid false positives, we take few QC steps done. To detect sequencing errors, polymerase chain reaction (PCR) duplication and adapter contaminations, several series of quality checks, the sequencing data quality, the proportion of GC contents, adaptor sequence residues, sequence overrepresented and duplication level, performed using FastQC. The remaining adapters were trimmed using cutadapt. Both fast QC and cutadapt were processed using qc analysis pipelined TrimGalore (https://github.com/FelixKrueger/TrimGalore), software. By following de facto single nucleotide polymorphism (SNP) and insertions and deletion (INDEL) calling GATK4 pipeline introduced by Broad Institution.
Preprocessed sequencing reads were aligned against human reference genome GRCh37 p.13 using Burrow Wheeler Aligner (BWA)’s maximal extract mathes algorithms. To correct duplication that occurs from the PCR stage, mapped reads were processed using GATK4 MarkDuplicate. then, base quality score recalibration was conducted to avoid exaggerated quality scores for each base. To discover short variants from individual recalibrated bam files, the GATK4 HaplotypeCaller module with the Genomic Variant Call Format (GVCF) model were performed. The GVCF files were generated for each individuals, the GVCF is a lossless format that contains every position whether the calling tool detects variants or not. Then each result was merged and processed for the ready-to-be joint genotyping status using the GenomicsDBimport module from GATK4. In order to detect and compare SNPs and INDELs in each individual, single VCF file contains 148 patients were jointly called using the GentoypeGVCFs module and separated into SNPs and INDELs by SelectVariants.
Filtering variants and annotation
Using the module variant quality score recalibration (VQSR) according to variants National Center for Biotechnology Information (NCBI)’s dbsnp151 from GATK4. A VQSR tranche sensitivity cutoff was applied to SNPs at 99.9 and 99% to indels as well as genotype calls with a phred-scaled quality score < 20 were also removed from variants callset for use in downstream analyses. To understand a more specific relationship between phenotype and genetic variations detected in the RP patients, we applied Snpeff originally supplied annotation GRCh37.75 downloaded from Snpeff as well as extra annotation files dbsnp151, dbnsfp4.1a which was obtained from NCBI and Jpopgen contains the detailed genomics coordinates of variants ID, gene, clinvar, HGSv, Polyphan using SnpSift.
Ethics statement
This study protocol was reviewed and approved by the Institutional Review Board (IRB) of Soonchunhyang University Hospital in 2017 (IRB No. 2017-12-031) to perform gene analysis with WES in inherited retinal disorders. Also, this retrospective study based on patients’ electronic medical records was approved by the IRB (IRB No. 2021-07-031) with waived informed consent and adhered to the tenets of the Declaration of Helsinki.
RESULTS
Ninety-four patients initially diagnosed with RP and sixty-four of their family members were enrolled in this study. Of the 158 subjects, six were suspected to have choroideremia based on fundus examination. Of these patients, five were confirmed to have CHM mutations and one was confirmed to have CNGB1 mutation by WES. The demographic and clinical features of these latter five subjects are summarized in Table 1.
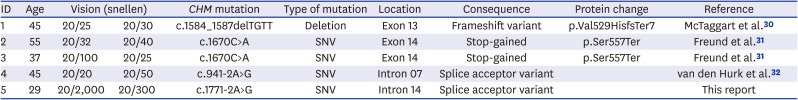
Table 1
Summary of clinical, genetic data of 5 patients with diagnosed choroideremia among patients initially diagnosed with retinitis pigmentosa
| ID | Age | Vision (snellen) | CHM mutation | Type of mutation | Location | Consequence | Protein change | Reference | |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 45 | 20/25 | 20/30 | c.1584_1587delTGTT | Deletion | Exon 13 | Frameshift variant | p.Val529HisfsTer7 | McTaggart et al.30 |
| 2 | 55 | 20/32 | 20/40 | c.1670C>A | SNV | Exon 14 | Stop-gained | p.Ser557Ter | Freund et al.31 |
| 3 | 37 | 20/100 | 20/25 | c.1670C>A | SNV | Exon 14 | Stop-gained | p.Ser557Ter | Freund et al.31 |
| 4 | 45 | 20/20 | 20/50 | c.941-2A>G | SNV | Intron 07 | Splice acceptor variant | van den Hurk et al.32 | |
| 5 | 29 | 20/2,000 | 20/300 | c.1771-2A>G | SNV | Intron 14 | Splice acceptor variant | This report | |
![]()
Hemizygous CHM gene mutation was observed in five patients; one frameshift (c.1584_1587delTGTT, p.Val529HisfsTer7), two nonsense (c.1670C>A, p.Ser557Ter), and two splice acceptor mutations (c.941-2A>G and c.1771-2A>G). The first four mutations have been reported previously, but the latter is novel.
Fundus examinations revealed significant atrophy of the choroid, diffuse yellow-whitish reflex of the sclera, and large choroid vessel with fine and granular pigmentary changes in the mid-peripheral fundus (Fig. 1). FAF showed small areas of hyperfluorescence in the macula and hypofluorescence due to the chorioretinal atrophy. In one case, hyperfluorescence was not observed in the macula. On FAG, except for the macula, hypofluorescence due to the loss of RPE and choriocapillaris was observed up to the far-peripheral retina (Fig. 2). OCT revealed relatively good preservation of the macula, but decreased outer nuclear layer (ONL) thickness, ellipsoid zone, and external limiting membrane attenuation, RPE loss, and choroidal thinning were observed. In addition, microcysts in the inner nuclear layer (INL), outer retinal tubulation (ORT), and wedge-shaped hyporeflective ONL structures (interlaminar bridges; ILBs) were observed in four patients (Fig. 3). The VF showed a small amount of remaining central field, and ERG showed no response.

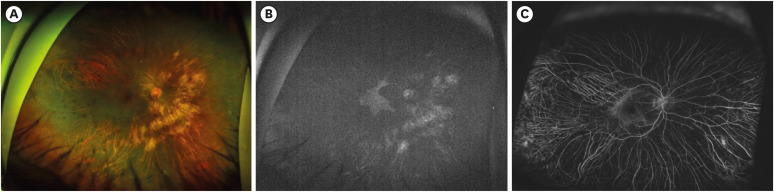

Fig. 1
Wide field color fundus photography of the right and left eyes of a patient with advanced choroideremia.
Wide field color fundus photographs show granular and lumpy material, chorioretinal atrophy, and a yellow-whitish sclera in peripheral and peripapillary regions. RPE and choroid remain in only a small area of the macula.
RPE = retinal pigment epithelium.
![]()
Fig. 2
Multimodal imaging of a male choroideremia patient. (A) Color fundus photography shows granular material in the mid-peripheral fundus, chorioretinal atrophy, and preservation of the posterior pole. (B) FAF showed a small area of hyperfluorescence in the posterior pole, in line with the results of color fundus photography shown in (A). (C) FAG revealed hypofluorescence up to the far-peripheral retina, with prominent choroidal vessels.
FAF = fundus autofluorescence, FAG = fluorescein angiography.
![]()
Fig. 3
Representative OCT images of choroideremia patients. OCT shows marked atrophy of the RPE, except in the macula, with intraretinal cysts, ORT (arrowhead), and ILB (arrow). Marked thinning of the ONLs was observed and the choroid was virtually absent, both of which are hallmarks of choroideremia.
OCT = optical coherence tomography, RPE = retinal pigment epithelium, ORT = outer retinal tubulation, ILB = interlaminar bridge, ONL = outer nuclear layer.
![]()
DISCUSSION
Choroideremia affects about 1 in 50,000 individuals and is caused by a genetic defect in one single gene called the CHM gene, which is located on the X-chromosome. The CHM mutation was reported in about 1% of patients with inherited retinal disorder in WES.1516 The pathogenetic mechanism of choroideremia has not yet to be fully elucidated, and the disease progresses very slowly, making early diagnosis difficult. Retinal thickening occurs in the earliest disease stage, although normal lamination is maintained. Over time, photoreceptor loss and RPE changes occur, and thinning of the retina eventually progresses. Although choroideremia is characterized by fundus findings, progressive VF loss, decreased ERG amplitude, and X-linked inheritance, it is often misdiagnosed as another inherited retinal disorder.17181920
Choroideremia and RP have several common features, such as night blindness, constriction of the VF, gradually reduced visual acuity, and retinal degeneration, and therefore the two conditions may be confused. Previous studies have suggested that about 6% of individuals diagnosed with RP-related disorders actually have choroideremia, while about one quarter of clinically diagnosed choroideremia cases may actually be other diseases, including RP.213141721 Choroideremia is characterized by scalloped chorioretinal atrophy beginning in the mid-peripheral fundus, with preservation of the macula. Considering the high variability in the appearance of the fundus in RP, choroideremia without a typical fundus appearance can easily be misdiagnosed as RP.
Bae et al.22 suggested that molecular genetic diagnosis would be helpful for proper diagnosis of choroidemia. In the present study, 5 (5.3%) of 94 subjects initially diagnosed with RP were shown to have CHM mutation by WES. However, one patient of 6 suspected choroideremia was confirmed as CNGB1 frameshift mutation (c.2670_2671dupGA), which has been known to be pathogenic in Japanese population. Therefore, genetic analysis and several imaging modalities, including OCT, FAF, and FAG, are required for diagnosing choroideremia.
Recently, several multimodal imaging methods, such as OCT, FAF, optical coherence tomography angiography, and scanning laser ophthalmoscopy, have been used to diagnose choroideremia. Khan et al.23 reported that INL microcysts, intraretinal edema, and ORT could be revealed by OCT. Jacobson et al.4 reported that photoreceptor layer and RPE degeneration, and ILBs, were found in these patients. In the present study, INL microcysts, intraretinal edema, and ORT were observed; ILBs were also seen along with inner segment/outer segment (IS/OS) loss. ORT is thought to occur at the boundary between disrupted outer retinal layers and a preserved photoreceptor layer. However, this finding is observed in several degenerative retinal disorders, i.e., is not specific to choroideremia.112425 ILBs at the boundary between normal and abnormal lamination were observed in all seven choroideremia patients reported by Lazow et al.,26 while Sun et al.27 reported ILBs in 10 of 12 choroideremia patients, and Syed et al.28 reported ILBs in male and female carriers. In the present study, ILBs were observed in four of five choroideremia patients. One patient with no ILBs had severe chorioretinal atrophy on OCT and almost no normal lamination was observed. To date, there have been no reports of ILBs in other degenerative disorders. Therefore, if ILBs are seen on OCT, choroideremia should be suspected.
FAF showed a clearer boundary between normal RPE and the lesion due to the accumulation of lipofuscin fluorophores. In particular, the Robson-holder ring, which is a hyper-autofluorescent ring observed at the IS/OS junction, as well as in the normal retina due to OS dysgenesis and lipofuscin production, is mainly observed in RP. Similarly, in this study, the Robson-holder ring was not observed in choroideremia patients, but was observed in those with RP. Furthermore, Popovic reported that the size of the ring correlates with visual function and follow-up imaging of the ring may help to predict disease progression and prognosis.29 FAF revealed a small area of preserved choriocapillaries and RPE in the central stellate region in choroideremia patients. The fundus and FAF images of five choroidemia patients representing these features were all shown in the Supplementary Fig. 1. FAF could be useful for diagnosing RP and choroideremia.
FAG can reveal loss of the choriocapillaries and preservation of choriocapillaries perfusion, and can also be used to observe choroidal neovascularization, but is rarely applied in choroideremia patients.1011 However, in this study, hypofluorescence caused by loss of RPE and choriocapillaries was observed in the entire retina, including the retinal and choroidal vessels. Although a “hypofluorescent background” was observed in both choroideremia and RP patients up to the mid-peripheral fundus, greater preservation of fluorescence was observed in the latter group. Therefore, FAG can be helpful for distinguishing between choroideremia and RP, which are difficult to distinguish based on visual examination alone.
In summary, of the 94 patients initially diagnosed with RP, 5 (5.3%) showed a CHM mutation by WES. Choroideremia may be misdiagnosed as RP due to their similar clinical manifestations. Choroideremia should be considered in the diagnosis of male patients with extensive choroidal atrophy. To improve the diagnosis of choroideremia, it is important to analyze patients using several imaging modalities, and to verify the diagnosis by genetic analysis.
 XML Download
XML Download