

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Hereditary spherocytosis (HS) is a common cause of congenital hemolytic anemia with an incidence rate of 1 in 2,000 Caucasians and 1 in 5,000 Koreans [1]. HS is caused by defects in red blood cell (RBC) membrane components, such as α-spectrin, β-spectrin, ankyrin, band 3, and protein 4.2, which are encoded by SPTA1, SPTB, ANK1, SLC4A1, and EPB42, respectively [2]. Spherocytosis, type 1 (OMIM #182900) caused by ankyrin defects is most common in Korea and is mostly inherited in an autosomal-dominant manner [1, 3].
HS is often suspected from hemolytic anemia with spherocytes on a peripheral blood smear (PBS) and the presence of a family history. However, its diagnosis is very difficult if the phenotype is mild and if there is no family history. Indeed, some cases have remained undiagnosed for decades [4, 5]. In addition, the low sensitivity and specificity of conventional tests such as PBS and osmotic fragility test (OFT) limit the diagnosis of HS [6]. Molecular diagnostic tests can overcome these limitations, which are especially useful in patients with a mild phenotype and no family history. Here, we report a case of HS that presented uncertain results from OFT but was diagnosed by a de novo ANK1 variant using multi-gene panel testing.
CASE
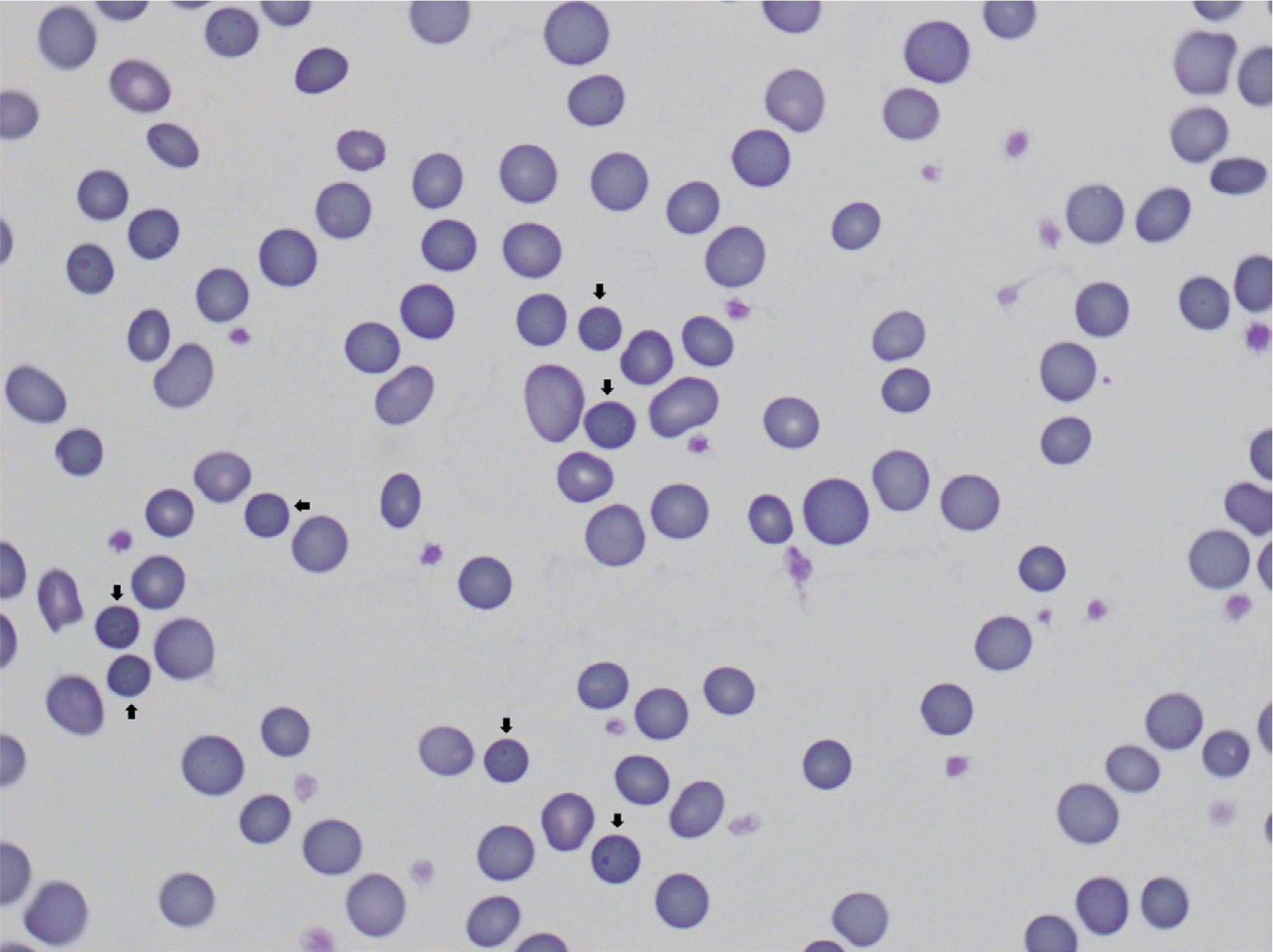
The patient was a 20-year-old woman without a family history of HS. She was born at 38 weeks of gestation and had a history of neonatal jaundice. At 3 months of age, she was transferred from a local clinic for bronchiolitis, sinusitis, and anemia. The initial complete blood count was as follows: Hb 8.4 g/dL (reference range: 11.0–15.0 g/dL); Hct 24.1% (34.3–49.9%); mean corpuscular volume 67.2 fL (80.0–97.7 fL); mean corpuscular hemoglobin concentration 34.7 g/dL (31.6–35.8 g/dL); reticulocyte 5.3% (0.5–1.5%). The corrected reticulocyte count was 2.8%. Total bilirubin was within normal range, but lactate dehydrogenase was elevated to 634 IU/L (100–225 IU/L), and haptoglobin was decreased to 36.2 mg/dL (55–324 mg/dL). The direct and indirect antiglobulin test results were negative. Hepatosplenomegaly was absent, and spherocytes were not observed in PBS. Two months later, the patient’s Hb level was 8.4 g/dL, with an increased reticulocyte count of 6.6% and urinalysis result of 1+ for urobilinogen. At the age of 18 months, her Hb level was 11.8 g/dL, with an increased reticulocyte count of 5.0%. The OFT was performed without RBC transfusions, and the results were within the normal range [hemolysis began 0.5%; completed 0.36% NaCl (0.5% and 0.35%)]. She visited our hospital again, at the age of nine, when she received pneumococcal and meningococcal vaccination. Hb was 9.7 g/dL, and the results of the OFT indicated increased fragility (began 0.64%; completed 0.48% NaCl). At the age of 16 years, the patient underwent a cholecystectomy for cholecystitis. In PBS, microcytic hypochromic anemia with spherocytosis was observed with a moderate/2+ grade (Fig. 1) [7]. Hb was 10.2 g/dL, and the OFT was within the normal range (began 0.52%; completed 0.36%). We did not perform an eosin-5′-maleimide (EMA) binding flow cytometry test.
When the patient was 20 years old, next-generation sequencing (NGS)-based multi-gene panel testing was conducted with the patient’s consent. The multi-gene panel included more than 2,155 probes targeting 85 genes associated with hematopathies. NGS was performed using the Illumina NextSeq 550Dx (Illumina, San Diego, CA, USA) with 150 bp paired-end sequencing. Alignment of sequence reads to the reference genome hg19, variant calling, and annotation were also conducted [8, 9].
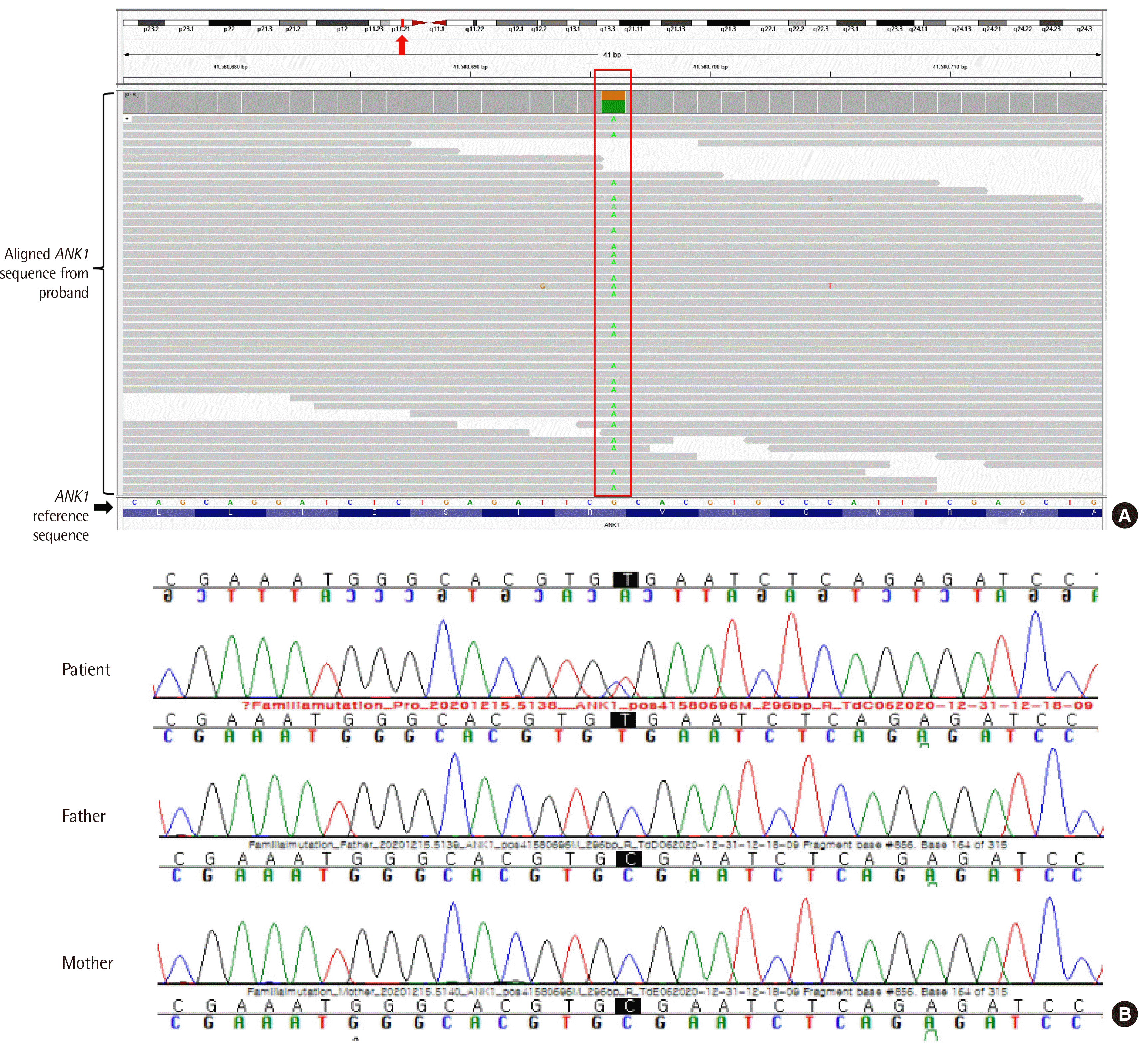
We identified a heterozygous variant, NM_000037.3(ANK1): c.856C>T (p.Arg286*), in exon 9 of ANK1. This variant was validated using Sanger sequencing and was classified as a pathogenic variant (PVS1+PM2+PP5) according to the 2015 American College of Medical Genetics and Genomics–Association for Molecular Pathology guidelines [10]. The patient’s parents did not carry this variant (Fig. 2). This study was approved by the institutional review board of our institute (IRB 2020-12-011). The patient has been prescribed folic acid and is being examined biannually at an anemia clinic without regular RBC transfusion or splenectomy.
DISCUSSION
HS is characterized by spherocytes in PBS. The defects in the RBC membrane components induce a reduction in the surface area, resulting in the formation of spherocytes. Spherocytes show increased osmotic fragility and are trapped and destroyed in the spleen. Destruction of spherocytes in the spleen leads to hemolytic anemia, jaundice, and splenomegaly [11]. Clinical manifestations vary from asymptomatic to severe. Undiagnosed HS may lead to complications such as gallstones and aplastic anemia. Complications may be an initial manifestation in patients with asymptomatic or mild phenotypes [12]. Therefore, early diagnosis is important to reduce the risk of complications [13].
HS is often misdiagnosed because conventional tests, such as PBS and OFT, have low sensitivity and specificity. Approximately 10% of HS patients have very few or no spherocytes in PBS [14]. The OFT results can also be normal in 10–20% of HS patients [15]. In addition, these tests are not specific because secondary spherocytosis cannot be distinguished. While the flow cytometry-based EMA binding test is the test of choice and current guidelines recommend its use for HS screening [15], it is not available in our laboratory, and we did not use it for our patient. Therefore, molecular genetic testing is very useful for diagnosis when the results of conventional tests are uncertain. According to a previous study, the results of PBS and OFT were normal in 8% and 13.2% of patients with genetic variants, respectively [1]. In our patient, spherocytes and splenomegaly were first observed at the age of 16 years. In addition, OFT was performed three times, but the results differed each time. Therefore, achieving a definite diagnosis is difficult with these traditional tests.
Approximately 75% of HS cases are inherited in an autosomal dominant manner, whereas both parents of the remaining HS cases do not display clinical symptoms and are often normal in hematological tests [16]. In these remaining patients, HS could be inherited in an autosomal recessive manner or developed due to a de novo mutation [16]. Autosomal dominant HS is associated with variants in ANK1, SLC4A1, and SPTB. Autosomal recessive HS is mostly associated with variants of SPTA1 and EPB42. Variants in ANK1 and SPTB are also associated with autosomal recessive HS. De novo HS is mostly associated with variants of ANK1 and SPTB [17]. In ANK1, approximately 15–20% of variants have been reported as de novo [18]. Identifying the inheritance pattern of HS is important for family planning and proper genetic counseling. However, in the absence of a family history, the inheritance pattern of HS cannot be identified without genetic testing. Specifically, when both parents are clinically and hematologically normal, it is impossible to distinguish whether HS is inherited in an autosomal recessive manner or occurs de novo, without genetic testing. Therefore, HS diagnosis with genetic testing is useful when conventional methods cannot confirm the disease.
In Korean patients with HS, ANK1 is the most common causative gene, followed by SPTB [3]. Between HS patients with ANK1 and those with SPTB variants, there were no significant differences in clinical characteristics except for the frequency of splenectomy. Splenectomy was more frequent in patients with ANK1 variants than in those with SPTB variants [3]. In patients with ANK1 variants, there were differences in Hb levels depending on the location of the variants. Patients harboring variants in the spectrin-binding domain exhibit more severe anemia than those harboring variants in the membrane protein binding domain or regulatory domain [3, 19]. The variant found in our patient was located in the membrane protein-binding domain. She did not have severe anemia, with an Hb level of 9.2±2.0 g/dL. Another case with an ANK1 p.Arg286* variant has also been reported to show a mild phenotype [19].
In our patient, findings of spherocytosis in PBS with an increased reticulocyte count, decreased haptoglobin levels, negative antiglobulin tests, and uncertain results of OFT were detected, but a definitive HS diagnosis was not reached without molecular genetic testing. Therefore, multi-gene panel testing, including HS-related genes, is a useful diagnostic tool in de novo cases. In patients with unexplained hemolytic anemia, molecular genetic testing should be considered for early diagnosis and proper management.
 XML Download
XML Download