

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Hemophilia is an X-linked blood disorder that causes sustained bleeding after injuries or trauma. Symptoms mainly include bleeding in the joints and muscles. The term hemophilia was first used by Friedrich Hopff, University of Zurich, in 1828 [1, 2]. Hemophilia B (HB) is caused by defects in coagulation factor IX. It is also called “Christmas disorder”, after it was identified by Stephen Christmas in 1952 [3]. FIX activity can be classified as severely (<1%), moderately (1–5%), and mildly (5–30%) impaired [1, 4, 5]. HB is less common than hemophilia A (HA) (1 in 25,000 males vs. 1 in 5,000 males worldwide) [6, 7]. Approximately 14,000 people with hemophilia have been registered at the Haemophilia Federation of India; however, hemophilia remains under-diagnosed and many cases are thus not registered. HA occurs in 1 out of 10,000 male births, while HB occurs in 1 out of 30,000 male births [8].
The F9 gene (Xq27.1-q2.2) is approximately 33.5 kb in length and contains eight exons [6, 9]. FIX is homologous to clotting factors VII (FVII) and XI (FXI). It plays a crucial role in blood coagulation and is synthesized in the liver as a vitamin K-dependent serine protease (SP) precursor [1]. FIX circulates in plasma as a glycoprotein. During blood coagulation, inactive FIX is converted into an active SP called Factor IXa. This activation process occurs in two ways: through intrinsic and extrinsic pathways. In the course of the intrinsic pathway, Factor IXa activates FIX in the presence of calcium ions. During the extrinsic pathway, FIX is activated by factor VIIa in the presence of calcium and lipoprotein [10]. FIX protein is composed of a γ-carboxyglutamic acid-rich (Gla) domain, two epidermal growth factor-like domains (EGF1 and EGF2), and an SP domain [6].
Recently, more than 3,000 pathogenic mutations and neutral polymorphisms have been identified in the F9 gene, and these mutations have been documented in various online hemophilia databases [6, 11, 12]. In this study, we analyzed the F9 genes of 150 HB patients from the hemophilia societies of Karnataka, India, for molecular changes using sequencing technology.
Go to : 
MATERIALS AND METHODS
Sample collection
This study was approved by the Institutional Ethical Committee of Shri B. M. Patil Medical College, Hospital and Research Centre, BLDE (Deemed to be University), Vijayapura (Ref. No.: BLDE (DU)/IEC/340/2018-19), and the SNMC Institutional Ethics Committee of Human Subject Research, Bagalkot (Ref. No.: SNMC/IECHSR/2018-19/A-B2/1.1). Written informed consent was obtained from all HB patients before blood sample collection. A total of 150 HB patients were included in this study, which were followed up at 12 different hemophilia societies across the Karnataka state of India. A detailed clinical history was obtained from all HB patients.
F9 gene analysis
Peripheral blood from HB patients was collected in EDTA vacutainers (BD, Franklin Lakes, NJ, USA). Prior to DNA isolation, the FIX concentration was measured and inhibitor assay were performed. DNA was extracted from peripheral blood using a blood and tissue DNA extraction kit (QIAGEN, Hilden, Germany). All exonic regions were amplified and the products were sequenced on an Applied Biosystems (ABI) 3500 Sanger sequencing platform using the BigDye Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific, Waltham, MA, USA). Results were analyzed using DNA sequence analysis software v5.4.
Bioinformatics analysis
The pathogenicity of the novel non-synonymous variants was analyzed using bioinformatics tools, such as PROVEAN (http://provean.jcvi.org/seq_submit.php), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/index.shtml), PHD SNP (https://snps.biofold.org/phd-snp/phd-snp.html), SNPs & GO (https://snps.biofold.org/snps-and-go/snps-and-go.html), PANTHER (http://www.pantherdb.org/), and SNAP2 (https://www.rostlab.org/services/snap/). The conservation property of missense variants was investigated using the Clustal Omega multiple sequence alignment tool (https://www.ebi.ac.uk/Tools/msa/clustalo/). A 3D model of wild type and mutant FIX proteins was predicted using the Swiss model (https://swissmodel.expasy.org/), and the results were visualized and analyzed using the UCSF Chimera program.
Go to :
RESULTS
Of the 150 HB patients included in this study, 102 (68%; FIX concentration, 0.6±0.2; age of onset, 2.0±1.0 y), 30 (20%; FIX concentration, 2.5±1.3; age of onset, 7.5±2.8 y), and 18 (12%; FIX concentration, 8.0±2.6; age of onset, 10.0±3.5y) suffered from severe, moderate, and mild HB, respectively. The detailed clinicopathological parameters are summarized in Table 1. In our study, we recorded 16 mutations. Of those, 1 was a synonymous mutation, 12 were missense mutations, 2 were stop-gain mutations, and 1 was a 3’ UTR variant. Notably, 13 (81.25%) mutations were previously reported, but 3 (18.75%) were novel mutations, which had not been entered into any of the human SNP databases. The majority of the mutations were found in exon 8 of the gene and largely comprised missense mutations. Exon 8 showed a high number of mutations compared to other exons of the F9 gene (Fig. 1, Table 2).
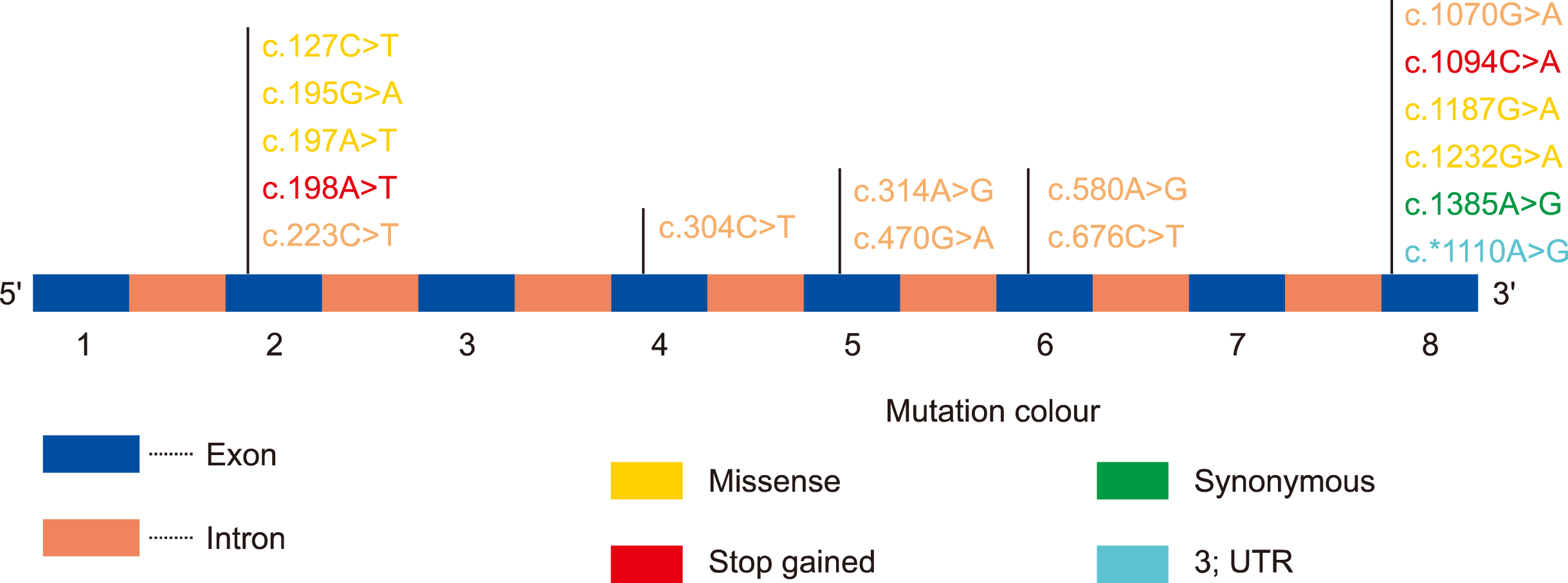
Table 1
Clinicopathological characteristics of the study population.
![]()
Table 2
List of mutations recorded in the F9 genes of our study cohort.
| Mutation type | Nucleotide change | cDNA position | Amino acid change | Exon | Status | Frequency, N (%) |
|---|---|---|---|---|---|---|
| Missense | g.11313C>T | c.127C>T | p.R43W | 2 | Reported (rs1603264205) | 9 (6.0%) |
| Missense | g.11381G>A | c.195G>A | p.M65I | 2 | Reported (rs763568424) | 1 (0.66%) |
| Missense | g.11383A>T | c.197A>T | p.E66V | 2 | Reported (CM940423) | 3 (2.0%) |
| Missense | g.11384A>T | c.198A>T | p.E66D | 2 | Not reported | 12 (8.0%) |
| Stop-gain | g.11409 C>T | c.223C>T | p.R75a) | 2 | Reported (rs137852227) | 2 (1.33%) |
| Missense | g.15369 T>C | c.304C>T | p.C102R | 4 | Reported (CM960574) | 2 (1.33%) |
| Missense | g.22664 T>C | c.314A>G | p.G143R | 5 | Reported (CM940499) | 4 (2.66%) |
| Missense | g.22706 G>A | c.470G>A | p.C157Y | 5 | Reported (rs1367198680) | 17 (11.33%) |
| Missense | g.25386 A>G | c.580A>G | p.T194A | 6 | Reported (rs6048) | 3 (2.0%) |
| Missense | g.25482C>T | c.676C>T | p.R226W | 6 | Reported (rs137852240) | 6 (4.0%) |
| Missense | g.36020 G>A | c.1070G>A | p.G357E | 8 | Reported (rs137852275) | 8 (5.33%) |
| Stop-gain | g.36044C>A | c.1094C>A | p.S365a) | 8 | Not reported | 15 (10.0%) |
| Missense | g.36137G>A | c.1187G>A | p.C396Y | 8 | Reported (rs137852273) | 1 (0.66%) |
| Missense | g.36182 G>A | c.1232G>A | p.S411N | 8 | Reported (rs137852276) | 3 (2.0%) |
| Synonymous | g.36335 A>G | c.1385A>G | p.Ter462= | 8 | Reported (rs561793582) | 1 (0.66%) |
| 3’ UTR | g.37446A>G | c.*1110A>G | ……… | 8 | Not reported | 3 (2.0%) |
![]()
The novel missense mutation c.198A>T was found in the F9 genes of 4 patients with moderate HB, with a mean FIX concentration of 2.0±0.5 and a mean age of onset of 7±1.0, as well as in 7 patients with severe HB, with a mean FIX concentration of 0.3±0.5 and a mean age of onset of 1.5±0.5. This mutation was observed at a high rate among patients with moderate HB. The novel stop-gain mutation c.1094C>A was found in 3 patients with moderate HB, with a mean FIX concentration of 2.6±0.8 and a mean age of onset of 6.5±0.5, as well as in 12 patients with severe HB, with a mean FIX concentration of 0.05±0.15 and a mean age of onset of 2±1.0. This mutation was observed at a high rate among patients with severe HB. The following, previously described pathogenic mutations have been observed: c.127C>T in 3 mild (FIX concentration, 8.1±1.9), 4 moderate (FIX concentration, 2.1±0.2), and 2 severe (FIX concentration, 0.65±0.15) cases; c.470G>A in 3 mild (FIX concentration, 7.6±2.7), 5 moderate (FIX concentration, 2.9±0.9), and 9 severe (FIX concentration, 0.1±0.7) cases; c.676C>T in 1 moderate and 5 severe (FIX concentration, 0.1±0.7) cases; c.1070G>A in 1 mild, 3 moderate (FIX concentration, 2.8±0.8), and 4 severe (FIX concentration, 0.05±0.1) cases; c.223C>T in 2 severe cases; and c.314A>G in 1 moderate and 3 severe (FIX concentration, 0.1±0.09) cases. The remaining mutations were recorded at a low frequency. Detailed genotype-phenotype associations are explained in Table 3.
Table 3
Genotypic and phenotypic associations of the mutations recorded in the study population.
![]()
The novel missense mutation p.E66D was shown to be harmful to FIX protein function by PROVEAN, SNAP2, PholyPhen2, SNP&GO, PHD-SNP, and PANTHER (Table 4). Multiple sequence alignment of this novel missense mutation indicated that it was present in the highly conserved residue of the FIX protein (Fig. 2). Homology modeling of the protein structure was conducted using the Swiss model server, which was then visualized and analyzed using chimera program. In p.E66D, the mutant residue is smaller than wild type residue, which might lead to the loss of interactions with the metal ion: “calcium 4 or magnesium 1; via 4-carboxyglutamate” (Fig. 3). The novel stop-gain mutation, p.S365*, results in a premature stop codon that leads to a truncated FIX variant of 365 amino acids, corresponding to a loss of approximately 21% of the wild type FIX protein (Fig. 3).
 | Fig. 2Multiple sequence analysis of the Factor IX (FIX) protein. The arrow indicates the position of the p.E66D mutation.
|
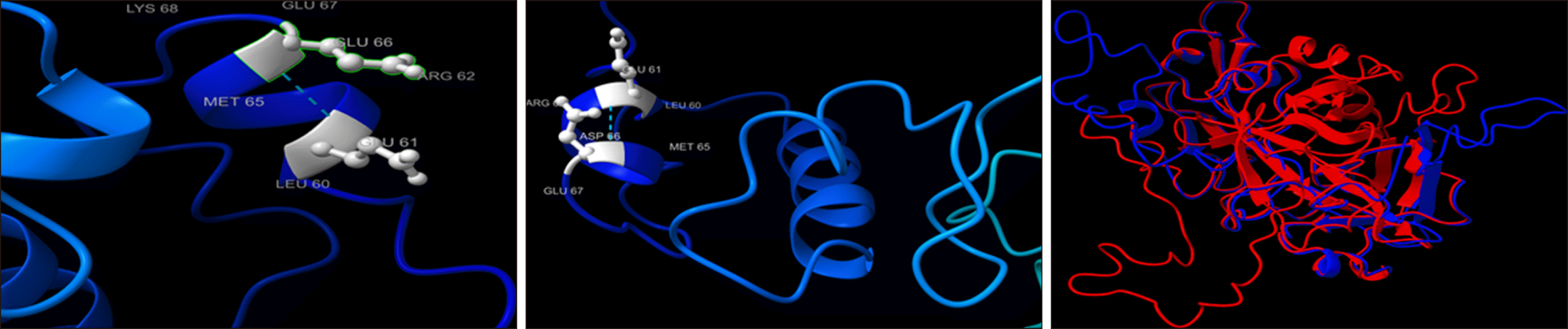 | Fig. 3Segments of a 3D model of wild type (upper left panel) and mutant (p.E66D; upper right panel) Factor IX. A superimposed 3D model of wild type (red) and mutant (p.S365*; blue) Factor IX are shown in the lower panel.
|
Table 4
Pathogenicity predictions of the p.E66D mutation.
| Mutation | PROVEANa) | SNAP2b) | PolyPhen2c) | PHD-SNPd) | SNP&GOe) | PANTHER |
|---|---|---|---|---|---|---|
| E66D |
Deleterious Score: -2.540 |
Effect Score: 37 |
Probably damaging Score: 0.999 |
Disease Score: 4 |
Disease Probability: 0.705 |
Probably damaging |
a)PROVEAN: “Deleterious” if the prediction score was ≤2.5, “Neutral” if the prediction score was ≥2.5. b)NAP2: “Neutral” if the score ranged from 0 to -100. “Effect” if the score was between 0 and 100. c)PolyPhen2: “Probably damaging” is the most disease-causing ability, with a score near 1. “Possibly damaging” signifies less disease-causing ability with a score of 0.5–0.8. “Benign”, which does not alter protein function, with a score closer to 0. d)PHD-SNP: if the probability is >0.5, mutation is predicted as “Disease” and if less than <0.5, mutation is predicted to be “Neutral”. e)SNP & GO: Probability of >0.5 is predicted to be a disease-causing nsSNP.
![]()
Go to :
DISCUSSION
HB is a bleeding disorder that causes abnormal or poor blood clotting. Pathogenic variants of the F9 gene are known to cause this disorder [13]. FIX is an essential element in the intermediate stage of the blood coagulation cascade. Nearly one third of hemophilia cases occur without any previous family history, but mainly due to new genetic variations [7]. In 1.3% to 7.8% of hemophilia cases, more than one mutation in the F9 gene have been reported [14], and, in accordance with other reports, most of the mutations were single nucleotide variants [15]. Missense mutations were the most common single nucleotide variants in HB, accounting for more than 58.4%, whereas 15.4% were frameshift mutations resulting from deletions, insertions, or duplications. Nonsense and splice mutations accounted for 8.3% and 9.4% of the cases, respectively [6].
In the present study, we recorded a total of 16 mutations, of which 15 (93.75%) were coding sequence variants. The majority of them (81.25%) were missense variants.
We recorded the stop-gain mutation p.R75W in 2 patients with severe HB, which have been described previously by Parrado Jara et al. (2020) [5], Radic et al. (2013) [16], Kwon et al. (2008) [17], Saini et al. (2015) [18], and Koeberl et al. (1990) [19]. This mutation is known to result in the loss of >80% of total FIX protein and to increase the probability of an immune response. The p.R226W mutation affects zymogen activation processes by altering the cleavage of Factor XIa [20, 21], which was recorded in 1 moderate and 5 severe (FIX concentration, 0.1±0.7) cases within our study group. The p.C396Y mutation was shown to disturb the environment of the active site that is involved in the conversion of the zymogen into an active enzyme, which was observed only in one severe case of HB [22]. We also recorded the p.T194A mutation in 3 mild HB cases (FIX concentration, 9.8±0.8) and the p.C357Y missense mutation, which were predicted to be detrimental to FIX protein function in the blood coagulation process in previous studies. We recorded the latter mutation in 1 mild, 3 moderate (FIX concentration, 2.8±0.8), and 4 severe (FIX concentration, 0.05±0.1) cases [23-26]. R43 is a mutation hotspot that accounted for 73.5% of HB cases in the present study [27], and we recorded the R43W mutation in c.127C>T in 3 mild (FIX concentration, 8.1±1.9), 4 moderate (FIX concentration, 2.1±0.2), and 2 severe (FIX concentration, 0.65±0.15) cases.
The novel missense mutation p.E66D was recorded in 4 patients with moderate HB, with a mean FIX concentration of 2.0±0.5, and in 7 patients with severe HB, with a mean FIX concentration of 0.3±0.5. It was found to be harmful to the function of the FIX protein by in silico pathogenicity prediction tools. Moreover, this novel mutation was found to be present in highly conserved regions of the FIX protein. The mutant p.E66D residue was found to be smaller than the wild type residue. This might have led to the loss of interactions with the metal ion: “calcium 4 or magnesium 1; via 4-carboxyglutamate”. Hence, this missense variant has detrimental effects on normal FIX activation and blood coagulation processes. According to the American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) guidelines, p.E66D is classified as “pathogenic” [28]. Along with missense variants, we also recorded one novel stop-gain mutation, p.S365*, in 3 moderate HB cases, with a mean FIX concentration of 2.6±0.8, as well as in 12 severe HB cases, with a mean FIX concentration of 0.05±0.15. This led to the loss of 21% of FIX protein, which resulted in the loss of most of the peptidase S1 activity of the FIX protein. According to the ACMG/AMP guidelines, p.S365* is classified as “pathogenic.” Only one 3’ UTR variant was recorded in our study cohort. Functional analysis of the mutations was the major limitation of our study.
Of the 150 HB patients, 90 (60%) had mutations in the F9 gene, including mild, moderate, and severe conditions. In the mild, moderate, and severe groups, 15 (10%), 26 (17%), and 49 (32%) of the patients showed mutations in the F9 gene. Among the 3 groups, the severe group showed a high rate of mutations (32%) compared to the other 2 groups; in our study cohort, the number of patients with severe HB was high, followed by moderate and mild groups (Table 1). Of the 16 mutations detected, 9 were identified as pathogenic. All of the 49 hemophilic patients with high mutation rates featured these 9 pathogenic variants: c.127C>T, c.198A>T, c.223C>T, c.314A>G, c.470G>A, c.676C>T, c.1070G>A, c.1094C>A, and c.1232G>A. These corresponded to the mutation locations causing the pathogenic effects that may have led to severe symptoms observed in hemophilic patients of our study population. Both c.1187G>A and c.1385A>G mutations were recorded as benign, and one moderately affected patient was diagnosed with the c.1385A>G variant, whereas the c.1187G>A mutation was found in a patient with severe HB. Two mutations, i.e., c.195G>A and c.304C>T, were likely pathogenic, and the c.195G>A variant was found in one patient with moderate hemophilia. In two patients with mild disease, the c.304C>T mutation was found. The likely benign mutations c.197A>T and c.580A>G were recorded in 2 mild, 1 severe, and 3 mild cases of hemophilia, respectively.
Additionally, 2 stop-gain mutations and 7 missense mutations were major pathogenic variations or disease-causing mutations recorded in our study; patients who were carriers of these mutations showed more severe conditions compared to the other 2 groups (mild and moderate). Likely pathogenic and benign mutations were also missense mutations, and all the patients carrying these mutations were mildly to moderately affected by HB, except for 2 patients. Only 3 mutations (c.127C>T, c.470G>A, and c.1070G>A) were associated with different severities. Moreover, 2 mutations were only associated with mild HB (c.304C>T and c.580A>G), 2 mutations were associated with moderate HB (c.195G>A and c.1385A>G), and 3 mutations were only associated with severe HB (c.223C>T, c.1187G>A, and c.1232G>A). One mutation each was associated with mild-moderate (c.*1110A>G) and mild-severe HB (c.197A>T). In addition, 4 mutations were associated with moderate-severe HB (c.314A>G, c.198A>T, c.676C>T, and c.1094C>A). Among the patients with severe HB, 48% (49/102) featured mutations in the F9 gene, followed by 86.6% (26/30) and 83.3% (15/18) of moderate and mild HB cases, respectively.
In our study, we also observed clinical differences between the mutated and wild type F9 groups. The main clinical symptoms were lower FIX concentration levels (5.5±2.5%) in the mutated groups compared to the control group (8.0±2.5%). The inhibitor-positive rate was also high in patients carrying F9 mutations compared to patients with wild type F9 (40/38), but inhibitor-negative numbers were decreased in the mutated hemophilic patients (46/112). Different studies conducted on HB patients recorded remarkable allelic heterogeneity, consisting of similar mutations, which were associated with different phenotypes. Similar to the results obtained by previous studies, we also found that the same mutations resulted in multiple phenotypes/severities [29]. Studies conducted between 1993 and 2014 with 20 or more unrelated patients with HB showed that the F9 detection rate varied from 83% to 100% using PCR and Sanger sequencing [4], whereas in our study population, the mutation rate was 60%. Phenotypic heterogeneity and mutation rates among HB patients are largely unknown. External influences, such as the modification of genes, environmental factors, or epigenetic effects, are major factors in both cases.
Our study strongly suggests that the majority of HB cases feature pathogenic single nucleotide variations, which may be novel or previously recorded. In many cases, novel single nucleotide variants are involved. Population-based screening of mutations will help establish inhibitor risk prediction and carrier detection strategies in India.
Go to :
 XML Download
XML Download