

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Juvenile dermatomyositis (JDM) is the most common idiopathic inflammatory myopathies (IIM) in children. It is a systemic capillary vasculopathy. Patients present with proximal muscle weakness, raised muscle enzymes, and pathognomic skin rashes such as heliotrope rash, Gottron’s papules. The course of JDM is variable. Author review some important advances in understading of pathogenesis, epidemiology, clinical manifestations, and treatment of JDM.
MAIN SUBJECTS
Epidemiology
JDM occurs in all nations in the world. The peak age at onset is between 4 and 10 years [1]. JDM is more common in girls than boys, with a ratio of 2:1 [2]. The incidence of JDM in the United States 3 per million children per year, which is similar to that in the United Kingdom (3.2 per million children per year) [3,4].
The race distribution of JDM as reported in the US register indicated a predominance of white non-Hispanics (65.1%), followed by Hispanics (14.2%) [3].
Pathogenesis
The pathogenesis of JDM is not clearly understood. It appears that children with genetic susceptibility to JDM (HLA-DQA1*0501, HLA-DQA*0301, HLA-DRB1*0301) may have exposure to environmental triggers such as infection [5].
Complement-mediated damage of vessels is a major mechanism, with activated complement inducing further cytokine release an vessel injury via the membrane attack complex.
The importance of type 1 interferon in JDM is becoming evident. An inflammatory cascade with type 1 interferon response leads to overexpression of major histocompatibility complex (MHC) class I and maturation of dendritic cells. Overexpression of MHC class I upregulates adhesion molecules, which influence migration of lymphocytes, leading to inflammatory infiltration of muscle [6].
Clinical manifestations
Symmetrical proximal muscle weakness is the hallmark of JDM. Weakness is often insidious and difficult to differentiate from fatigue at onset. The weakness probably affects all muscle groups but is most obvious in the shoulder girdle, hip flexors, and neck flexors. Patients may have difficulty raising arms, combing hair, climbing stairs, and getting out of bed. Findings on examination include the Gower sign and Trendelenburg sign. Each reflecting proximal lower extremity weakness and gluteal muscle weakness. The affected muscles may be tender, edematous.
Weakness of the palatal and pharyngeal muscles manifests clinically by dysphonia, dysphagia, nasal speech, and regurgitation of liquids through the nose. Respiratory muscle weakness can be a medical emergency and lead to respiratory failure.
Arthralgia or arthritis are often seen in early phase of disease course. The arthritis is nonerosive and frequently involves the knees [7].
Pathognomonic rash include heliotrope rash, Gottron’s papules, periungual erythema with capillary loop abnormalities. The heliotrope rash is a symmetrical erythematous to violaceous rash of the upper eyelids, often accompanied periorbital edema. Facial erythema crossing the nasolabial folds is common, in contrast to the malar rash of systemic lupus erythematosus. Gottron’s papules are an erythematous, shiny, thickened plaques over the dorsal surfaces of the metacarpophalangeal, proximal interphalangeal joints and occasionally on the elbows, knees, ankle malleoli. JDM patients often exhibit photosensitivity to ultraviolet light exposure with erythema in sun-exposed areas. If seen over the posterior neck, upper back, shoulder, this erythema is known as the ”shawl sign”, and anterior neck, upper chest, this erythema is known as the “V sign”.
Evidence of small vessel vasculopathy is often visible in the nailfolds as individual capillary loops that are dilated, tortuous, or dropout. Nailfold capillary abnormalities may be observed in the clinic using a magnifier such as ophthalmoscope. Patients with active disease have a greater degree of nailfold capillary abnormalities compared with those with inactive disease [8]. Severe vasculopathy causes ulcerative skin lesions on fingers, arms, toes.
Calcinosis is reported in up to 30% of JDM patients [9]. Calcinosis reflects a dystrophic deposition of the calcium phosphate, hydroxyapatite in the skin, soft tissue. Calcific lesions tend to be at pressure points, such as buttocks, knees, and elbows. Calcinosis is thought to be associated with a delayed diagnosis, untreated disease, a chronic course, and inadequate therapy.
Pulmonary involvement in JDM is rare, but serious complication. Pulmonary diseases include interstitial lung disease, aspiration pneumonia secondary to pharyngeal and esophageal dysmotility, and alveolar hypoventilation secondary to respiratory muscle weakness.
Vasculopathy of the gastrointestinal (GI) tract develop in severe JDM, with severe abdominal pain, ulceration, hemorrhage, perforation, and infarction. Clinically important GI involvement is rare, but it is one of the causes of death in JDM.
Diagnosis
The diagnosis of JDM is made through Bohan and Peter [10] criteria published in 1975. This criteria include the following findings: classic rash (heliotrope rash, Gottron’s papule), proximal muscle weakness, elevation of the serum level of one of the muscle enzymes, electromyography (EMG) demonstrating denervation and myopathy, and a muscle biopsy consistent with inflammatory myosistis. According to the 1975 criteria, a diagnosis is considered definite if a patient presents with 3 criteria in addition to the classic rash, or probable if a patient presents with 2 criteria plus the rash (Table 1).
Because of their invasive nature muscle biopsy and EMG are no longer used as frequently. In practice, magnetic resonance imaging (MRI) is currently widely used to diagnosis of JDM, although it is not part of the Bohan and Peter criteria [11].
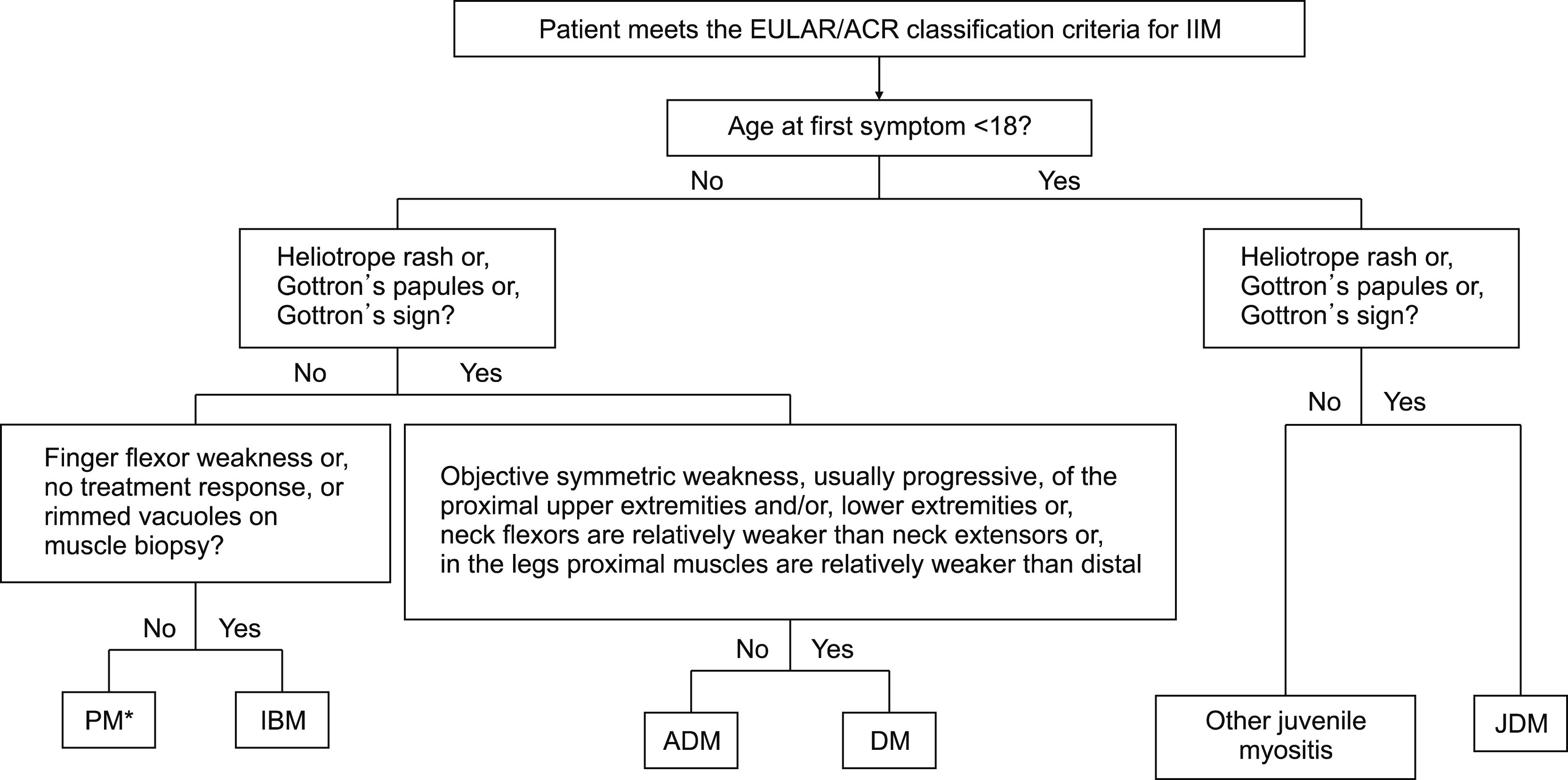
In 2017, the newly developed European League Against Rheumatism/American College of Rheumatology (EULAR/ ACR) classification criteria were published [12]. This criteria demonstrated high sensitivity and specificity. The criteria provide a score that is calculated from age of onset, variables of muscle weakness, skin manifestations, other clinical manifestations, laboratory measurements, and muscle biopsy findings. Because fewer pediatric patients undergo muscle biopsies, there is an additional scoring algorithm that does not include biopsy variables. Definite IIM corresponds to a probability of 90% or greater (total score of ≥7.5 without muscle biopsy or ≥8.7 with muscle biopsy). A total score of 5.5 or greater to less than 7.5 without biopsy or 6.7 or more to less than 8.7 with biopsy is classified as probable IIM, with a probability of 55% or more to less than 90%. A total score of less than 5.3 without biopsy or less than 6.5 with biopsy is classified as possible IIM with a probability of 50% or more to less than 55% (Table 2). A patient classified with IIM by the EULAR/ACR classification criteria (probability of IIM ≥55%) can be further subclassified with a classification tree (Figure 1).
Laboratory findings
Elevations in a number of muscle-derived enzymes, including creatine kinase, lactate dehydrogenase, aspartate aminotransferase, alanine aminotransferase, aldolase, are seen in JDM. LDH is better at reflecting the global activity and predicting relapse of disease. Erythrocyte sedimentation rate and C-reactive protein, may be elevated or normal. Antinuclear antibody is positive in >80% of JDM, and rheumatoid factor is typically negative [13].
Myositis-specific antibodies are exclusively found in children with IIM, and not in other autoimmune conditions such as juvenile idiopathic arthritis, systemic lupus erythematosus.
Patients with antitranscription intermediary factor 1 gamma antibodies tend to have cutaneous ulceration and show typical skin features such as Gottron papules, shawl sign, and need more aggressive treatment. Patients with 3-hydroxy-3-methylglutaryl-coenzyme A reductase present with profound weakness and have only a partial response to immuno-suppressive medications. Intravenous immunoglobulin (IVIG) seems to be effective for these children. Antinuclear matrix protein 2 antibodies is shown to be associated with younger age at disease onset, muscle cramps, dysphonia, a greater degree of muscle weakness, and calcinosis. Patients with antisignal recognition peptide antibodies have a high frequency of severe muscle weakness and cardiac abnormalities. Anti-Mi-2 antibodies were strongly associated with edema and a greater degree of weakness, malar rash, Gottron papules, and heliotrope rash. Patients with these antibodies usually have high antinulclear abtibody (ANA) titers [14].
MRI aid both diagnosis and monitor disease activity. T1-weighted images of the thigh demonstrate muscle atrophy and fatty infiltration in the presence of chronic disease. T2-weighted images with fat suppression demonstrate soft tissue edema and active inflammation. Short tau inversion recovery images improve visualization of inflammatory change.
Treatment
The goals of treatment are to control inflammatory myositis and prevent disease complication (e.g., calcinosis and contratures). There are no adequate controlled trials in the management of JDM. Thus treatments are based on clinical experience. Early, aggressive treatment of JDM associated with a better prognosis. Corticosteroids in combination with another immunosuppressive medication (most commonly methotrexate) is the mainstay of treatment.
Traditionally, standard treatment for milder cases has been high-dose oral prednisone at 2 mg/kg/day (maximum dose 60 mg daily), which is continued until clinical and laboratory improvement are evident and then slowly tapered over at least 2-year period [15]. In more severe cases with dysphagia, dysphonia, pulmonary involvement, high-dose intravenous methylprednisolone (30 mg/kg/day, maximum dose 1 g daily) is used. With corticosteroids treatment, however, most patients suffer adverse effects, including cushingoid appearance, cataracts, osteoporosis, and growth retardation. As a result, many JDM patients are now treated with adjunctive immuno-suppressant in an attempt to spare corticosteroids.
Oral or subcutaneous methotrexate (15 mg/m2 per week, maximum 40 mg) is widely used as a corticosteroid-sparing agent in JDM. Patients given methotrexate (15 mg/m2 per week orally or subcutaneously) at disease onset had a lower cumulative prednisone dose, less weight gain and improved height velocity, but achieved the same disease control as the comparison group who were treated only with prednisone [16]. Adverse events of methotrexate include immunosuppression, hepatitis, nausea, vomiting, and teratogenicity. Folic acid is typically given with methotrexate starting at a dose of 1 mg daily to reduce side effects of folate inhibition (anemia, oral ulcers).
Hydroxychloroquine is used as a secondary disease- modifying agent to reduce cutaneous disease and maintain remission. The recommendation dose is 5 mg/kg per day orally in either tablet or liquid form.
IVIG is now widely used as an adjunctive therapy for corticosteroid-resistant patients, especially resistant skin rash. The recommendation regimen is IVIG at 2 g/kg per infusion (maximum 70 g), with single infusion administered every 2 weeks for 5 doses, followed by monthly infusions for 2 years.
Cyclosporine is now most often considered in patients who are intolerant to methotrexate. The starting dose of cyclosporine is 3 to 5 mg/kg per day. Adverse events of cyclosporine are hypertrichosis, hypertension, hirsutism, and abdominal pain [17].
Recently, the efficacy on skin, muscle and global disease activity of cyclophosphamide has been reported in 56 severe and refractory cases of JDM [18]. The cyclophosphamide regimen in this study was 500 mg/m2 (maximum 500 mg) administered intravenously every 2 weeks for the first 3 doses, followed by 750 mg/m2 (maximum 1.2 g) every 3 to 4 weeks (according to the response, for a total of 6 to 7 doses).
A retrospective study of 50 JDM patients who received Mycophenolate Mofetil (MMF) as adjunctive treatment demonstrated a decrease in disease activity of both skin and muscle inflammation at 12-month follow-up [19]. Infection was the most common side effect and patients should be monitored for this complication. MMF is often used for patients who have methotrexate intolerance and persistent skin disease.
Anti-B-cell therapy with rituximab has been suggested as being potentially useful in treating JDM. Rituximab, anti-CD20 monoclonal antibody, has been reported to improve disease activity in a small case series of four JDM patients [20]. Reports of the use of other biological agents such as tumor necrosis factor (TNF)-α inhibitors, abatacept, tofacitinib are based on case reports with mixed results.
Other treatments including systemic tacrolimus, tofacitinib for severe disease have shown some merit in refractory cases.
Physical therapy might improve muscle strength and prevent contracture [21]. Bed rest is not indicated. JDM patients should apply photoprotective agents daily, even in winter and on cloudy days. Calcium and vitamin D are often given to reduce osteoporosis and osteopenia.
There is no standardized treatment of JDM-associated calcinosis. Current knowledge of treatment outcomes is confined to small series and case reports. Treatments were separated into two categories: immunomodulatory and alternative agents, the latter including drugs with non-immunosuppressive actions, such as altering calcium and phosphorus metabolism. Of the immuno-modulatory agents, the most frequently used include IVIG, systemic glucocorticoids and methotrexate. Less frequently used agents include TNF-α inhibitors, rituximab, and abatacept. For alternative agents, bisphosphonates were the most frequently used, followed by calcium channel blockers, intravenous/topical thiosulfate, aluminum hydroxide and warfarin. Bisphosphonates inhibit calcium turnover and remodeling [22]. Surgical removal should be considered in cases where the lesions cause significant limitation in mobility or significant pain
Clinical assessment
Muscle strength should be assessed at diagnosis and serially during follow-up. This is often done using Manual muscle testing of the standardized 8 muscles (MMT8) or Childhood Myositis Assessment Scale (CMAS). MMT8 assess 8 proximal (deltoid, biceps, gluteus maximus, gluteus medius, quadriceps), distal (wrist extensors, ankle dorsiflexors), and axial (neck flexors) muscle groups. MMT8 has been used as a primary end point for JDM clinical trials and in clinics follow patients progress. CMAS is a observational performance-based instrument of 14 functional tasks to assess muscle endurance, muscle function, strength. CMAS may be more informative but takes 15 minutes to perform [23].
Complications
JDM patients with acute and severe weakness are at risk for aspiration pneumonia and respiratory failure. Crampy abdominal pain and occult GI bleeding may indicate bowel wall vasculitis and lead to ischemia.
Lipodystrophy manifests in 14%∼25% of patients with JDM. This is characterized by a progressive, and symmetrical loss of subcutaneous fat. Lipodystrophy has been associated with insulin resistance, dyslipidemia, and type 2 diabetes.
Prognosis
Before the introduction of corticosteroid treatment, prognosis in JDM is very poor, with one third of the patients recovering without complications, one third developing disability, and one third dying. After the introduction of corticosteroid treatment, the mortality rate dropped to 1%.
At 7 years of follow-up, 75% of patients have little to no residual disability, but 25% continue to have chronic muscle weakness and 40% have chronic rash.
 XML Download
XML Download