

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
von Hippel–Lindau (VHL) disease is a multisystem cancer syndrome that is caused by variants of the VHL tumor suppressor gene (VHL) located on the short arm of chromosome 3 [1]. VHL encodes pVHL, a tumor suppressor protein that plays an important role in the regulation of hypoxia through the transcription factor, hypoxia-inducible factor (HIF) [2, 3]. In patients with VHL disease, multiple and/or bilateral tumors develop at a relatively young age and affect the central nervous system (CNS), kidneys, adrenals, pancreas, and reproductive organs [4, 5]. Although these tumors are benign, with the exception of renal cell carcinoma (RCC), VHL patients have the lowest life expectancy compared with patients with other tumor syndromes, such as neurofibromatosis and familial adenomatous polyposis [6]. The most frequent causes of VHL-related death are metastasized RCC and neurological damage due to CNS lesions [7-9]. Although several promising treatments for VHL disease are available, early detection of tumors is the first priority to manage the disease and has been shown to reduce the frequency of complications and mortality [6, 9-12].
There are a few reports on how VHL disease is managed according to current guidelines, such as the VHL Alliance guidelines [13], but most of these studies have been performed in western populations [7, 9, 14].
We report comprehensive clinical characteristics of Korean VHL disease patients, including age of onset and incidence rate of manifestations, and correlate these with VHL variant types. We set up a VHL clinic for VHL surveillance and genetic counseling and compared long-term data between VHL clinic participants and non-participants. To the best of our knowledge, this is the first study to present the clinical characteristics of VHL patients with surveillance performance rates that demonstrate the usefulness of a VHL clinic with integrated VHL-related departments to conduct surveillance and genetic counseling, in an Asian population.
MATERIALS AND METHODS
Population and procedures
Between May 2011 and May 2018, 54 patients attended the VHL clinic and were diagnosed as having VHL disease based on clinical diagnostic criteria of the Samsung Medical Center (SMC; Seoul, Korea) [1, 15]. Among the 54 patients, two patients passed away due to RCC-associated complications and two patients moved to another center during the study period. For the remaining 50 VHL patients, medical records were reviewed retrospectively. Clinical characteristics, including frequency of each manifestation, age at onset of each manifestation, rate of first onset of each manifestation, and genotype–phenotype correlations, were reviewed and molecular study results were collected after obtaining informed consent. The levels of hormones, such as catecholamine and metanephrine, were evaluated using plasma and urine (spot and/or 24-hour urine) tests.
Each clinical manifestation was categorized based on location as either CNS- or visceral organ-related manifestation. CNS lesions include retinal hemangioblastoma (R-HB), endolymphatic sac tumors (ELSTs), and craniospinal hemangioblastoma (CS-HB). All hemangioblastomas (HBs) located in the cerebral cortex, cerebellum, brainstem, or spinal cord were counted as CS-HBs. Other lesions, except CNS lesions, were included as visceral organ manifestations, including RCC, pancreatic neuroendocrine tumor (PNET), and epididymal cystadenoma. Clinical phenotypes were classified as type 1 or type 2 based on the presence of pheochromocytoma (PCC) [5, 16]. To monitor VHL surveillance, we classified the patients into three groups: group 1, patients who attended the VHL clinic once a year; group 2, patients who attended the VHL clinic once, but did not participate in annual follow-ups; and group 3, patients who were not registered at the VHL clinic, but who visited only the specialists relevant to their clinical manifestations. For patients in group 3, medical records and test results were retrieved through interdepartmental cooperation. The study protocol was approved by the SMC Ethics Review Committee (Institutional Review Board no. 2019-09-072).
Organization of the VHL clinic and surveillance program
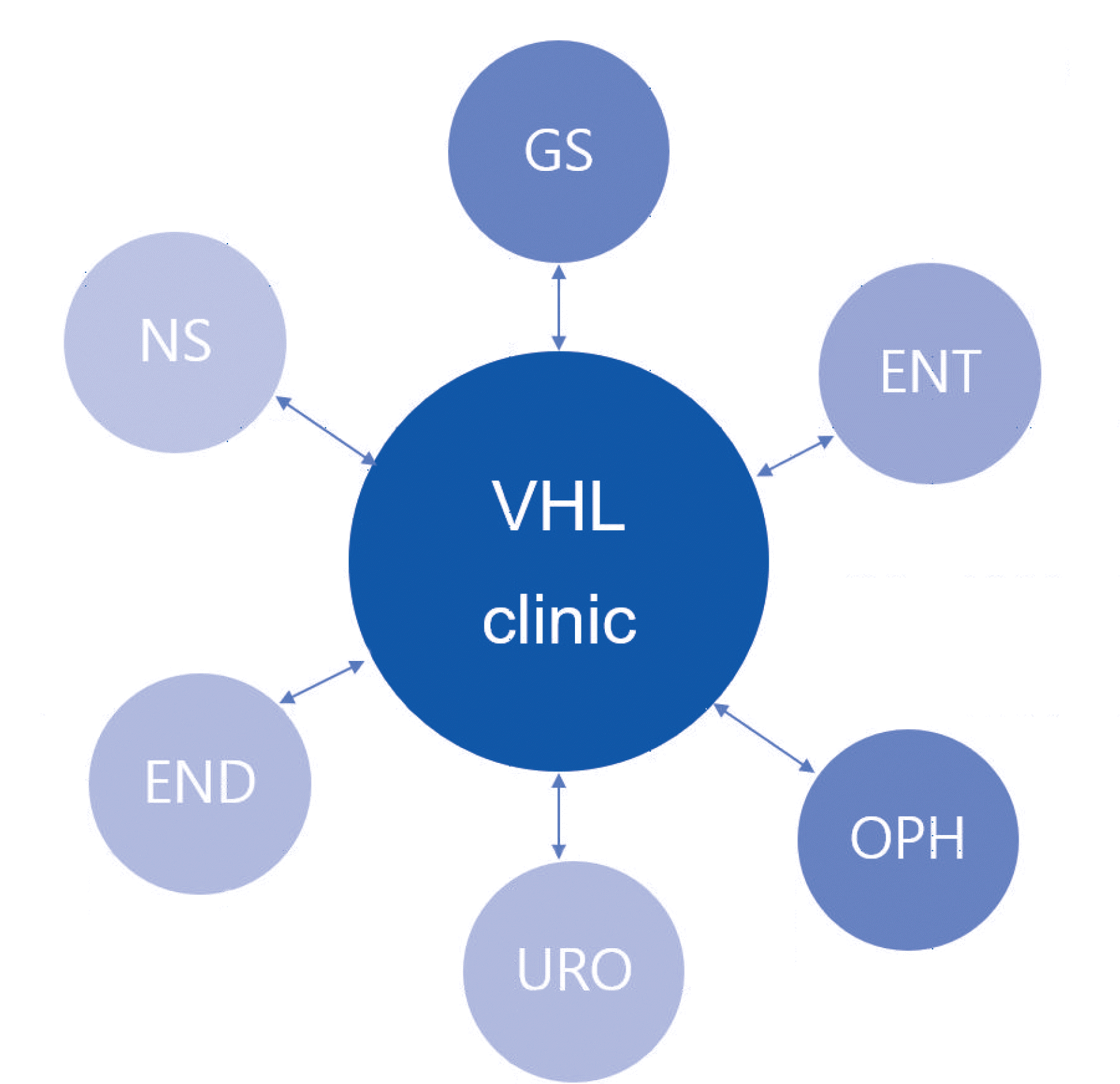
We recruited a multidisciplinary team, including an endocrinologist, urologist, general surgeon, neurosurgeon, ophthalmologist, otolaryngologist, and radiologist. In the VHL clinic, we performed VHL molecular analysis and executed surveillance with genetic counseling as per the VHL surveillance guidelines of the SMC. The VHL surveillance guidelines of the SMC include recommendations from the US National Institutes of Health and the Danish VHL Coordination Group [1, 9]. The surveillance comprised the following five major procedures: ophthalmic examination; CNS imaging using computed tomography (CT) or magnetic resonance imaging (MRI); CT, MRI or ultrasonography (US) of the abdomen; determination of catecholamine, metanephrine, nor-metanephrine, and vanillylmandelic acid (VMA) levels in 24 hour-collected urine or plasma; and an audiometry test. Each surveillance procedure was recommended to be performed yearly, beginning at the age of 15 years.
When new clinical manifestations of VHL disease were identified during surveillance, the patient was referred to the appropriate specialist (Fig. 1). Genetic counseling included educating the patient on VHL disease, including inheritance patterns, risk to offspring, and importance of surveillance, and recommendations for VHL screening of other family members.
VHL variant analysis
Blood samples were collected from the patients after obtaining informed consent. Genomic DNA was isolated from peripheral blood leukocytes using a Wizard Genomic DNA Purification Kit (Promega, Madison, WI, USA) according to the manufacturer’s instructions. Direct sequencing of the exonic and flanking intragenic regions of all three VHL exons was performed using an ABI 3130XL Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) and a BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems). Chromatograms were analyzed using Sequencer software version 5.0 (Gene Codes, Ann Arbor, MI, USA). If a pathogenic variant was not identified by direct sequencing, multiplex ligation-dependent probe amplification (MLPA) analysis (SALSA MLPA Probemix P016 VHL; MRC Holland, Amsterdam, The Netherlands) was performed to detect large genome rearrangements. The MLPA results were analyzed using the GeneMarker software (SoftGenetics, State College, PA, USA). Variants of VHL were classified into missense, nonsense, splicing, small insertion/deletion, or large deletion types. Genetic variants were interpreted as pathogenic or likely pathogenic as per the guidelines of the American College of Medical Genetics and Genomics [17].
Statistics
Data are expressed as number, percentage, median, or range and were analyzed using MedCalc version 15.5 (MedCalc, Ostend, Belgium) and Microsoft Excel 2010 (Microsoft, Redmond, WA, USA). To compare surveillance attendance, group 1 was considered the main analysis group and comparisons with groups 2 and 3 were made using the Fisher’s exact test. All reported P-values are two-tailed and P<0.05 was considered significant.
RESULTS
Patient demographics and clinical manifestations
Of the 50 patients included in the study, 15 (30%) were women, and the median age at onset of VHL disease was 33 years (range: 14–59 years). The median age at first symptom onset was 28 years (range: 14–54 years). The most frequent manifestation was CS-HB (76%, 38/50) and the second most common manifestation was RCC (72%, 36/50). The most frequent manifestation at first onset was CS-HB (46%, 23/50), and the median age at onset of CS-HB was 24 years (range: 16–43 years). Thirty-eight patients (76%) had type 1 VHL and 12 patients (24%) had type 2 VHL. Other demographics of VHL disease are summarized in Table 1.
Table 1
Clinical characteristics of VHL disease
![]()
Among the 23 patients who had CS-HB as a first manifestation, 16 (72%) had symptomatic manifestations, such as blurred vision and high blood pressure that required CT or MRI, compared with seven out of nine patients (85.7%) who had R-HB as a first manifestation. None of the patients (0%, 0/9) who had RCC as a first manifestation were symptomatic, and in 34 out of 36 patients (94.5%) who had RCC or renal cysts as a first manifestation, symptoms were identified incidentally by surveillance. In the case of PNET or pancreatic cysts, surveillance identified cysts in 19 out of the 21 patients. Other clinical characteristics of VHL disease, such as the percentage of symptomatic manifestations that led to first surveillance, are summarized in Table 1.
Genotype–phenotype correlations
Missense variants were the most common VHL variant type, followed by splicing variants in both the type 1 and type 2 groups (Fig. 2). Nonsense variants and small insertions/deletions were not observed in VHL type 2, although one large deletion was found. Approximately 16% of the VHL variants were large deletions that required MLPA analysis. Our findings were consistent with those reported in previous studies on variant frequencies or genotype–phenotype correlations in Asian populations [18-21].
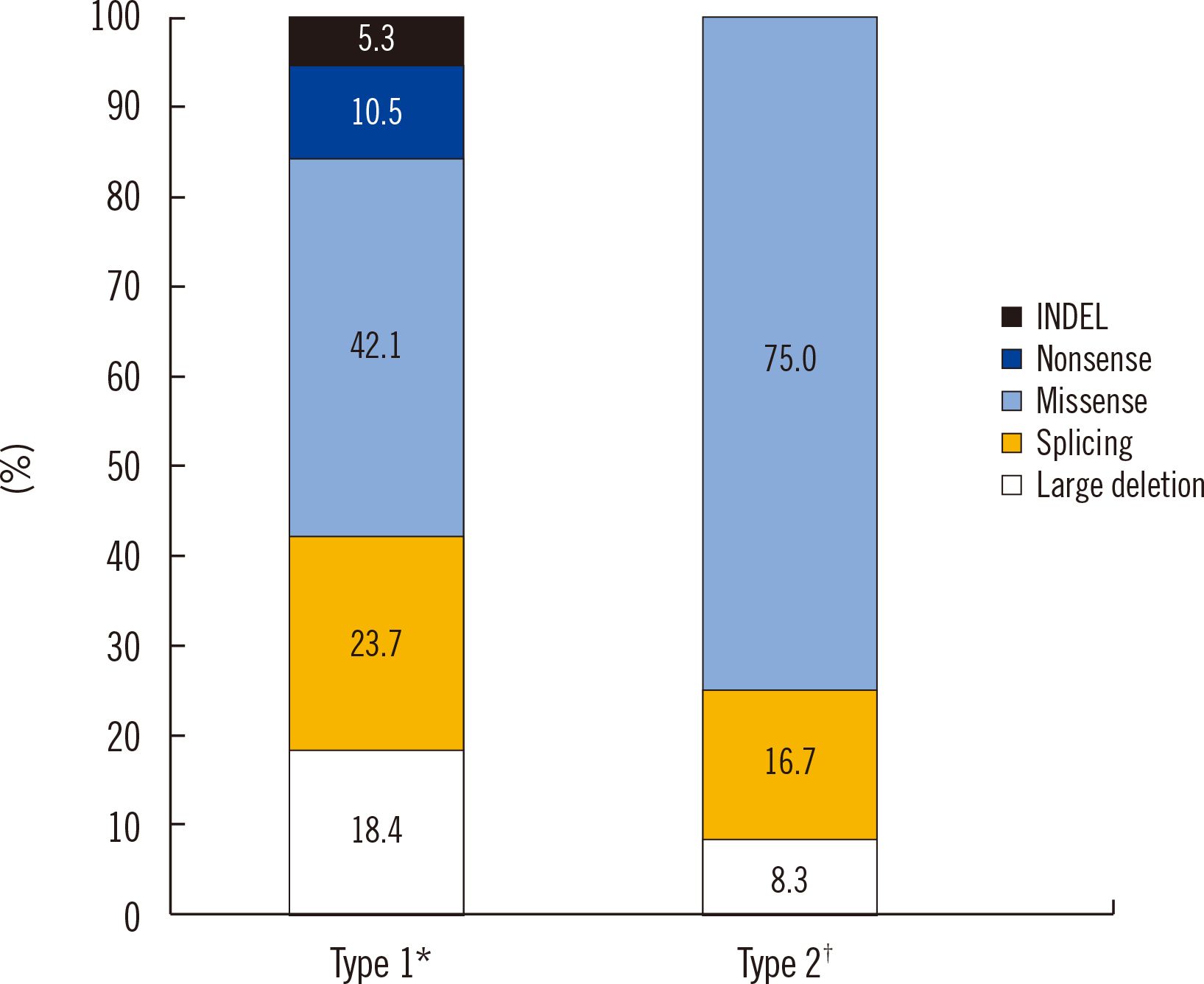
Performance rate of VHL surveillance
We assessed the performance rate of each surveillance test category (Fig. 3). Fifty percent of the patients (25/50) had registered at the VHL clinic. In group 1, all surveillance tests, except the audiometry test (94.4%, 17/18), were conducted once every 12 months (median; range: 2–48 months) (Supplemental Data Table S1). In group 2, catecholamine level determination and ophthalmic examination were conducted less frequently than in group 1 (71.4%, 5/7 vs. 100%, 18/18), although the difference was not significant (P=0.012). The performance rate of the audiometry test was lower in group 2 than in group 1 (94.4% vs. 42.9%, P<0.001). In group 3, the performance rates of the other surveillance tests were notably lower than those in group 1. Genetic counseling aimed at educating VHL patients regarding the inheritance patterns of VHL disease and recommendations for genetic tests for other family members were conducted in only 36% (9/25) of the patients in group 3.
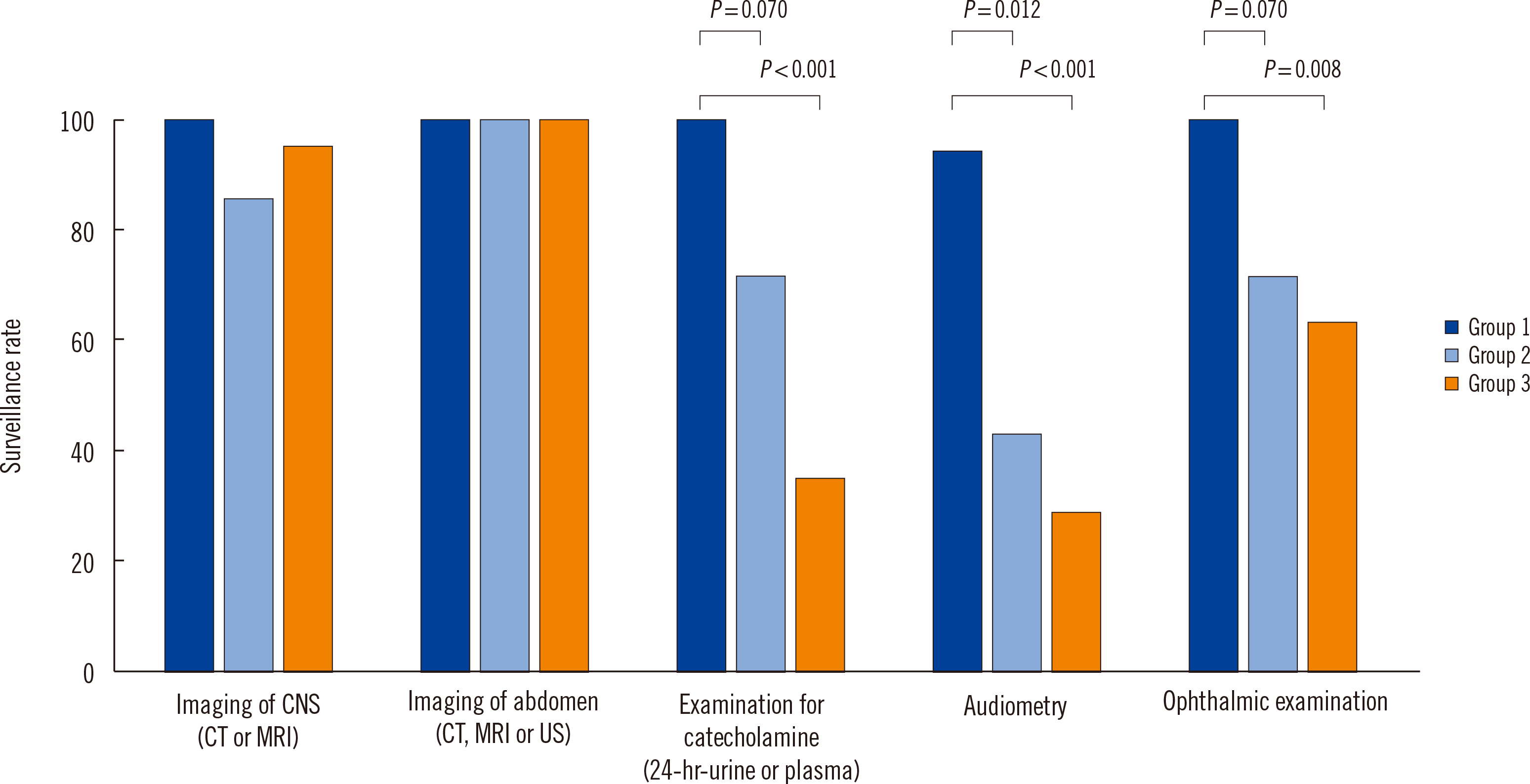
Fig. 3
Performance rates of surveillance tests in group 1 (patients who attended the VHL clinic once a year), group 2 (patients who attended the VHL clinic once, but did not participate in annual follow-ups), and group 3 (patients who were not registered at the VHL clinic, but who visited only the specialists relevant to their clinical manifestations).
Abbreviations: CNS, central nervous system; CT, computed tomography; MRI, magnetic resonance imaging; US, ultrasonography.
![]()
DISCUSSION
We reviewed the clinical characteristics of VHL disease and the performance rate of VHL surveillance attendance in a Korean population. Although clinical characteristics, such as the frequency of manifestations in patients and age at disease onset, were not distinctively different from those reported in other studies, RCC manifested more frequently than reported in other studies and the age at onset of CS-HB and RCC was lower than in other studied population groups [5, 9, 14, 16]. Most patients with CS-HB (69.6%, 16/23) or R-HB (77.8%, 7/9) as a first manifestation were symptomatic, which led to the first surveillance. In contrast, patients with visceral organ lesions, such as RCC (0%, 0/9), PCC (16.7%, 1/6), and PNET (0%, 0/2), were asymptomatic when these characteristics were observed as a first manifestation.
In most cases, visceral tumors, including RCC (94.5%, 34/36), PCC (80.0%, 12/15), and PNET (90.5%, 19/21), were diagnosed incidentally during surveillance. Therefore, regular surveillance according to the VHL Alliance guidelines is important for the early detection of VHL and especially, visceral organ lesions [13]. Both physicians and patients need to be aware of the possibility of asymptomatic manifestations and, in their own best interest, patients should allow the surveillance of visceral organs, such as the kidneys, adrenal glands, and pancreas.
Like patients in the VHL clinic participant group (group 1), patients in groups 2 and 3, who did not continue yearly follow-ups or did not enroll at the clinic, respectively, regularly received CNS imaging by CT or MRI as well as imaging of the abdomen by CT, MRI, or US (100% vs. 85.7% and 76.0%, respectively). These results show that the patients adhered well to scheduled inspections. However, the performance rates of ophthalmic examination, determination of pheochromocytoma-related hormones (catecholamine, metanephrine, nor-metanephrine, and VMA), and audiometry test were significantly lower in groups 2 and 3. These results suggest that patients do not come to the hospital on a regular basis when they do not experience symptoms.
Only nine patients in group 3 (36.0%, 9/25) received genetic counseling on the importance of surveillance and recommendations for the screening of other family members. Regardless of participation in the VHL clinic, the surveillance interval was not significantly different among patients if they had received each category of surveillance (Supplemental Data Table S1). This implies that physicians tend to be concerned only about their own specialty and that patients only visit the departments relevant to their symptoms. Patients received genetic counseling and were educated on the importance of surveillance if they visited the VHL clinic once, even if they did not participate in annual follow-ups at the VHL clinic. This may explain why no significant differences in the surveillance test performance rates were observed, except for the audiometry test, in patients who attended the VHL clinic, but did not participate in annual follow-ups.
Phenotypic heterogeneity is a hallmark of VHL disease; even within a family harboring the same variant, symptoms are variable [15]. Therefore, comprehensive multidisciplinary surveillance of multiple organs is required to screen for clinical manifestations associated with VHL disease. Surveillance guidelines for VHL disease have been recommended by several VHL study groups [1, 22, 23]. In addition, VHL is inheritable in an autosomal dominant manner and genetic counseling, including education on the inheritance pattern, potential risk to offspring, and reproductive options with family investigation, is required for VHL patients [16]. Although surveillance with genetic counseling is important to detect and manage VHL disease, it is challenging for a physician to conduct all surveillance measures in a timely manner while also providing the patient with comprehensive education [16]. In this respect, a VHL clinic with a multidisciplinary team is extremely useful for conducting comprehensive surveillance for VHL patients. VHL patients require lifelong, continuous management, which can be achieved in a single-center clinic with the multidisciplinary approach suggested here.
 XML Download
XML Download