

 PDF
PDF Citation
Citation Print
Print


Understanding the molecular biology of cancer has led to the development of targeted therapies that selectively attack specific genes and proteins that contribute to the growth and proliferation of certain cancer cells, thereby reducing harm to normal cells. Targeted therapies have brought about many changes in cancer treatment and have dramatically increased the survival rate of patient.
Tyrosine kinase inhibitors (TKIs) have been developed as effective targeted therapeutic agents to suppress abnormal signaling pathways triggered by the uncontrolled activation of cellular and receptor tyrosine kinases. With the approval of imatinib mesylate by the United States Food and Drug Administration in 2001 as the first TKI chemotherapeutic agent that inhibits the break-point cluster region-Abelson leukemia (BCR-ABL) fusion protein to treat Philadelphia chromosome-positive chronic myeloid leukemia (CML), BCR-ABL TKIs have contributed to outstanding advances in the treatment of this disease. Thereafter, there has been a continuous development of various TKIs, such as nilotinib, dasatinib, and bosutinib.1
Nilotinib, which is structurally similar to imatinib, is a second-generation BCR-ABL TKI. This drug inhibits BCR-ABL, discoidin domain receptor (DDR), stem cell factor receptor (KIT), and platelet-derived growth factor receptor (PDGFR) tyrosine kinases with 10–30 times more potent effects than imatinib. Thus, nilotinib has been used in patients resistant or intolerant to imatinib.2
Most TKIs have been reported to induce minor to moderate side effects, including headache, myalgia, neutropenia, thrombocytopenia, and gastrointestinal discomfort with nausea, vomiting, and diarrhea, compared to conventional cytotoxic drugs.1,3 Although there are a few cases of acute kidney injury, including tubular toxicity, tumor lysis syndrome, rhabdomyolysis in treatment with imatinib, renal injury associated with nilotinib has rarely been reported.3
Herein, we describe a case of AIN in a 39-year-old man with CML who had been treated with nilotinib.
CASE
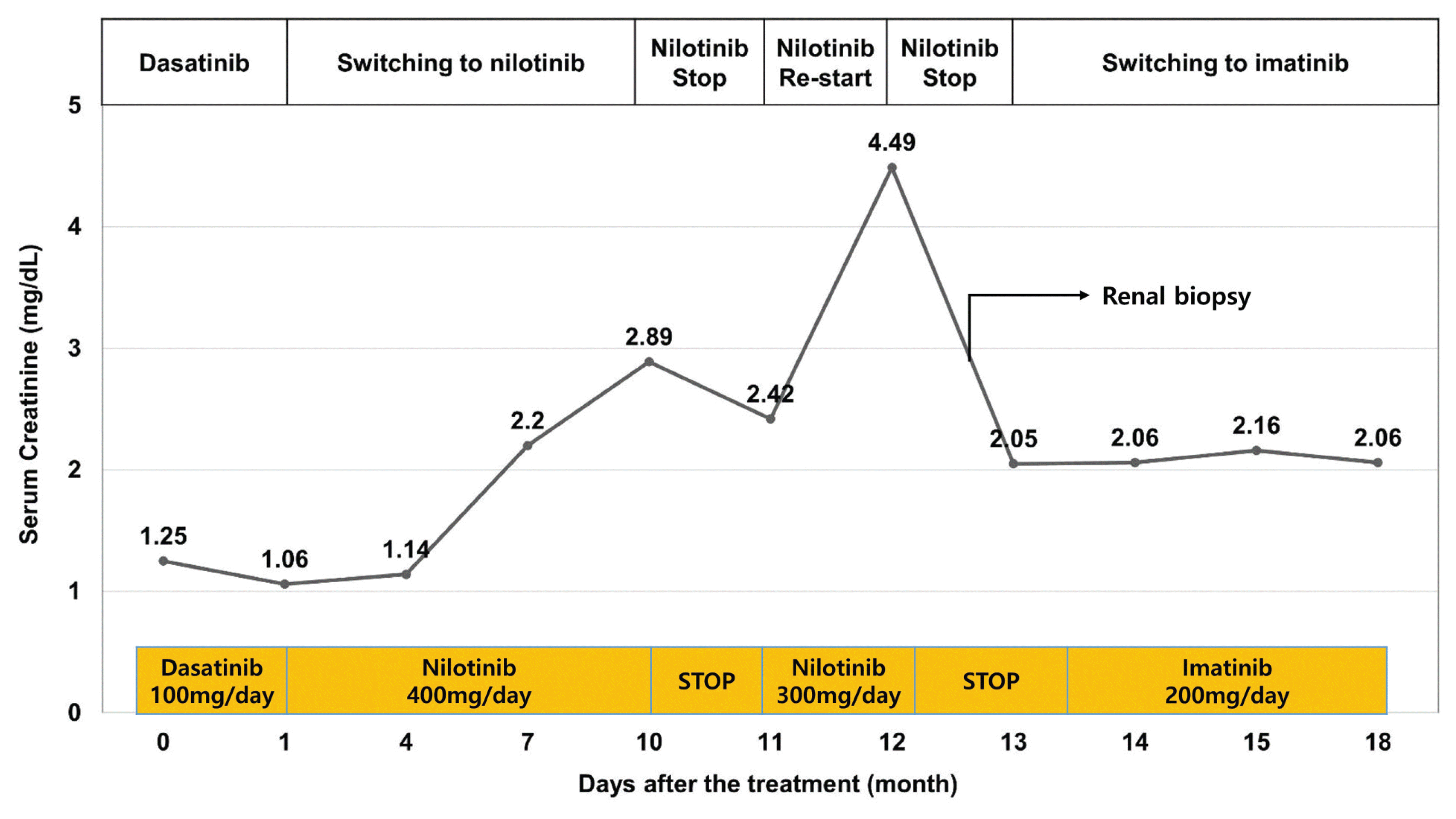
A 39-year-old man with chronic-phase CML was referred to the nephrology clinic due to acute renal injury. The patient started treatment with nilotinib (800 mg/day) 4 months before the first visit to the nephrology clinic. The first line of defense for the patient in terms of medication was dasatinib. However, this was changed to nilotinib due to marrow suppression and poor overall response. The changes in serum creatinine levels after nilotinib treatment are shown in Figure 1. After initiating nilotinib treatment, the serum creatinine level was 1.06 mg/dL and glomerular filtration rate was 83.1 mL/min/1.73 m2. Proteinuria and hematuria were not observed. Four months after the start of treatment with nilotinib, the patient showed a good response with a bcr-abl1 major real-time quantitative polymerase chain reaction (IS-NCN) of 3.99%. Nine months after the start of treatment with nilotinib, the patient presented with neutropenia and acute kidney injury (serum creatinine 2.89 mg/dL). One month after discontinuation of nilotinib, renal function improved (serum creatinine 2.42 mg/dL), but it did not return to the baseline level of renal function before treatment for CML. The patient restarted nilotinib (400 mg/day) for treating CML. However, the patient presented with a rapid elevation of the serum creatinine level (to 4.80 mg/dL). Chemotherapy was terminated once again, and the patient was admitted to a nephrology clinic to identify the cause of the acute renal injury. The patient had no history of comorbidities and medication causing renal injury.
On admission, his vital signs were as follows: blood pressure, 120/80 mmHg; pulse rate, 78/min; respiration rate, 20/min; and body temperature, 36.2 °C. Chest auscultation revealed no abnormalities. No significant findings were observed during abdominal examination, and no edema was found in the lower limbs. Blood test results were as follows: white blood cell count, 4100/mm3; hemoglobin, 9.7 g/dL; platelet count, 272 × 103/μL; sodium, 142 mEq/L; potassium, 4.3 mEq/L; chlorine, 112 mEq/L; blood urea nitrogen, 33.7 mg/dL; creatinine, 4.80 mg/dL; total protein, 7.4 g/dL; albumin, 4.3 g/dL; uric acid, 8.7 mg/dL; lactate dehydrogenase, 363 U/L; aspartate transaminase/alanine transaminase ratio, 10/8 IU/L; and C-reactive protein, 0.04 mg/dL. Additionally, IgA level was 146 mg/dL, IgG 1651 mg/dL, IgM 78 mg/dL, ASO (−), C3 119 mg/dL, C4 42 mg/dL, Fluorescent antinuclear antibody (−), and anti-dsDNA (−). Monoclonal gammopathy was not observed on serum or urine electrophoresis. The results of the urine test were as follows: specific gravity, 1.0111; pH, 5.5; urine protein (trace); spot urine protein/creatinine ratio, 250.1 mg/g; urine glucose (−); albuminuria (−); urine occult blood 3+; urine white blood cell count, 0–2/high power field; and urine red blood cell count, 21–29/ high-power field. Chest radiography revealed no significant findings. Abdominal ultrasound revealed normal-sized kidneys on both sides, with no significant findings (Fig. 2).
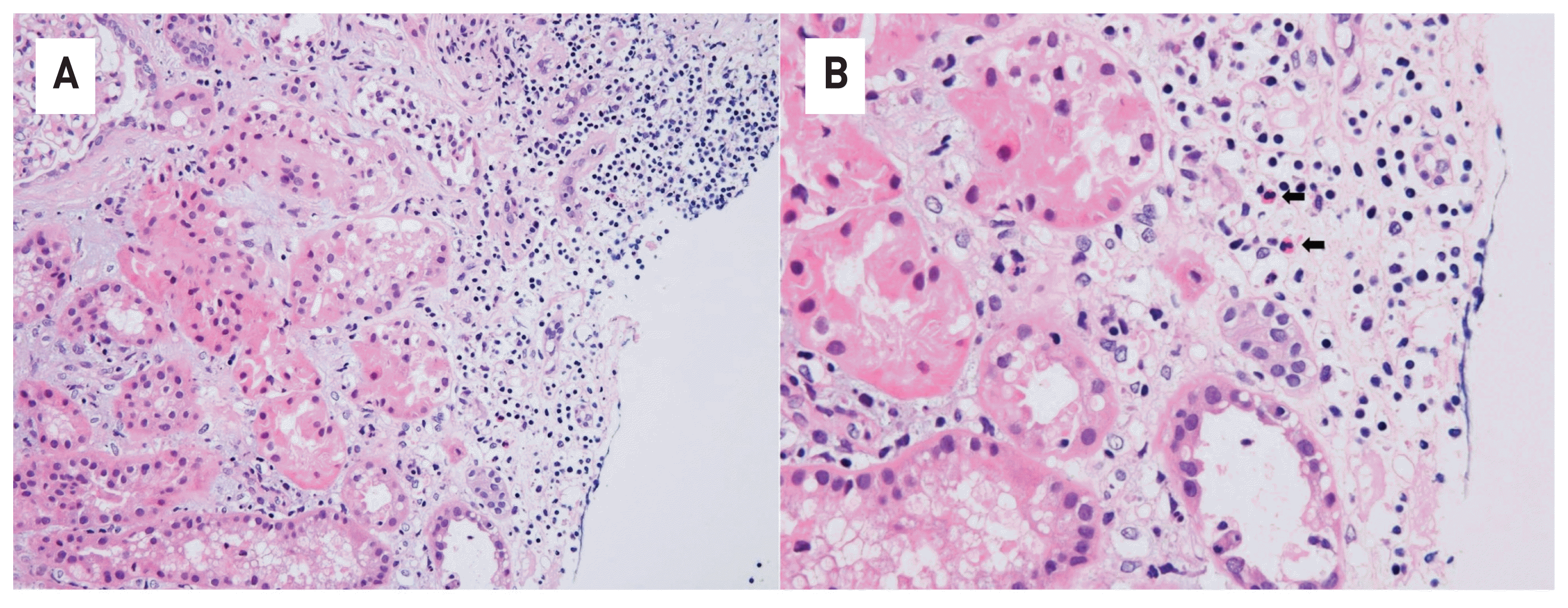
A renal biopsy showed infiltration of lymphocytes and eosinophils at the renal interstitium and peritubular area along with tubular necrosis (Fig. 3). No infiltration of immune complex deposits was observed by immunofluorescence microscopy. Electron microscopy revealed foot process effacement, but abnormalities in the glomerular baseline membrane and subendothelial deposits were not observed.
With the diagnosis of nilotinib-induced AIN, nilotinib was discontinued and intravenous fluid hydration was continued. Subsequently, the patient showed improved renal function after 3 months (serum creatinine level 2.05 mg/dL). Imatinib mesylate 100 mg/day was initiated for CML treatment. The dose of imatinib was gradually increased to 300 mg/day, and the renal function of the patient remained stable without further deterioration.
DISCUSSION
TKIs have shown remarkable survival benefits in patients with CML, with fewer adverse effects. Four TKIs, namely imatinib, nilotinib, dasatinib, and bosutinib, have been approved for the treatment of CML by the US Federal Drug Administration (FDA) and the European Medicine Agency (EMA) as first-line choices.1 Nephrotoxicity is a rare side effect of TKI treatments, particularly nilotinib.3 A few studies have shown that imatinib has a higher prevalence of renal injury than nilotinib or dasatinib. Yilmaz et al. investigated the estimated glomerular filtration rate change in 466 newly diagnosed chronic phase CML patients treated with imatinib, dasatinib, or nilotinib for 12 months. They reported that there was a decline in the mean glomerular filtration rate only among patients treated with imatinib and dasatinib and not in those treated with nilotinib. 4 Ren et al. analyzed the incidence of chronic renal injury in CML patients treated with imatinib, nilotinib, or dasatinib. The authors found a higher incidence of chronic renal injury in patients treated with imatinib than in those treated with nilotinib or dasatinib.5 Marcolino et al. demonstrated that imatinib treatment duration is related to a decreased estimated glomerular filtration rate in patients with CML.6
The mechanism of TKI-related renal injury is unknown. Imatinib can induce renal injury, which has been suggested to be associated with tumor lysis syndrome and tubular toxicity.3,4 Imatinib-induced tumor lysis syndrome causes acute renal injury that results from the precipitation of intracellular contents, such as uric acid and phosphate, released by rapid destruction of cells during cancer treatment of renal tubules.6 Tubular toxicity induced by imatinib was related to the PDGF-mediated pathway. Imatinib inhibits the PDGF-mediated pathway, which is critical for tubulogenesis after tubular injury.3
Dasatinib is known to be related to proteinuria, rather than decreased renal function. A phase I dose-escalation and a pharmacokinetic study reported that 18% of the patients treated with dasatinib showed proteinuria.7 The mechanism of proteinuria related to dasatinib has been associated with the disruption of vascular endothelial growth factor (VEGF) signaling, which involves podocytes and endothelial cells in humans.8
Nilotinib is metabolized mainly by cytochrome P450 3A4, and metabolites are excreted via the bile into feces and not into the urine. No dosage adjustment is needed in patients with renal impairment. 1,4 Renal dysfunction is a rare adverse reaction that develops during nilotinib treatment. None of the patients who participated in phase I and II trials of nilotinib for CML developed renal failure.7 To date, there have been only three case reports of nilotinib-induced renal injury, which were associated with tumor lysis syndrome in chronic phase CML patients.9 However, AIN from nilotinib treatment has not been described in the literature yet.
AIN is a cause of acute renal injury. Drug-mediated interstitial nephritis accounts for 70%–90% of all interstitial nephritis cases. It has histological characteristics including inflammation and edema of the renal interstitium.10 Concerning TKI-induced interstitial nephritis, VEGF-targeting drugs such as sunitinib and sorafenib have been described in some case reports. Sunitinib- and sorafenib-induced interstitial nephritis is suggested to be linked to blocking the VEGF pathways that mediate a variety of signaling pathways.11
In the present report, it is presumed that nilotinib might induce interstitial nephritis by blocking the BCR ABL. However, we believe that further research is needed to clarify the mechanism of nilotinib-induced interstitial nephritis.
Recently, immune-related adverse effects have been reported in patients with immune checkpoint inhibitors (ICPIs) as a new type of chemotherapy line. ICPIs are monoclonal antibodies used for cancer therapy by blocking inhibitory receptors expressed on T cells. Various antibodies against cytotoxic T lymphocyte-associated antigen 4 (CTLA-4), programmed death 1 protein (PD-1), and programmed death-ligand 1 (PD-L1) currently have been developed. Anti- CTLA-4 receptor antibodies promote the activity and proliferation of quiescent T-cells while anti-PD-1 receptor and anti-PD-L1 antibodies inhibit suppression of activated T-cells. The vigorous activated immune system is related to several immune- related adverse effects including nephrotoxicity. ICPI-associated acute kidney injury (AKI) has similar clinical and histological characteristics to other drug-induced acute interstitial nephritis, but with a long latency period. CPI-associated AKI might be related to a distinct mechanism of reprogramming of the immune system, through loss of tolerance with reactivation of drug-specific T cells induced by the immune checkpoint inhibitors.11 In case series of CPI-induced AKI, CPI discontinuation with corticosteroids initiation led to complete or partial improvement of the kidney.12 PD-1 receptor antibody-associated AKI can be induced by down-regulation of tolerance to the drug known to cause acute interstitial nephritis.13 CPI-associated AKI occurs relatively later than other drug-induced AKI. In one retrospective single-center study, the majority pathological finding of patients with CPI-associated AKI was acute interstitial nephritis. However it can frequently cooccur with other glomerular pathologies.14 A case series by Izzedine et al also reported that acute tubular injury, acute interstitial nephritis, and minimal change disease are the most common histological findings in renal injury associated with pembrolizumab, PD-1 receptor antibody.15 Because CPI-associated AKI lacks ideal biomarkers to distinguish it from other drug-induced AKI, renal biopsy is needed to identify the correct diagnosis.14
In this case study, we report a case of biopsyproven interstitial nephritis in a 39-year-old patient with CML who was treated with a second-generation TKI (nilotinib). Our patient had microscopic hematuria without any manifestations of hypersensitivity, such as skin rash, eosinophilia, and fever. The renal function of the patient decreased during treatment with nilotinib but improved when nilotinib was discontinued due to neutropenic fever. However, the renal function of the patient deteriorated again with the reintroduction of nilotinib for CML treatment. Through kidney biopsy, we found that the cause of the decreased renal function was AIN. The patient had not taken any offending drugs that commonly result in drug-induced interstitial nephritis, except for nilotinib. Finally, we diagnosed the patient with nilotinib-induced AIN. After switching to imatinib mesylate, the patient’s renal function stabilized without further deterioration.
Kidney injury associated with nilotinib can be underestimated because it is relatively uncommon. However, our case indicates that nilotinib can be a potential cause of renal dysfunction by inducing interstitial nephritis when renal function deteriorates following treatment with nilotinib. In patients diagnosed with nilotinib-induced interstitial nephritis, replacement with other TKIs could be considered to prevent further renal dysfunction, as in our case.
 XML Download
XML Download