

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Vascular calcification (VC) is defined as mineral deposition in the vasculature in the form of calcium-phosphate complexes. Although VC is regarded as part of the normal aging process, certain pathological processes, such as diabetes, hypertension, chronic kidney disease (CKD), and rare hereditary disorders may precipitate the condition.1) VC causes a loss of arterial elasticity, increased pulse pressure and systolic blood pressure, and left ventricular hypertrophy, and the severity of VC is closely associated with an increased risk of cardiovascular morbidity and mortality.2)
VC has been observed more frequently in adults with CKD than in age-matched general population,3) and a similar trend is observed in younger CKD patients.4) VC in CKD patients was initially considered to occur only in end-stage renal disease; however, recent studies have shown that VC is also observed in the early stage of CKD.5) Considering that cardiovascular disease is the most common cause of death in CKD patients, it is important to understand the pathophysiology of VC in CKD patients to detect early signs of VC and prepare for appropriate management.6) In this review, we present a review of the mechanism, diagnostic imaging modalities, clinical manifestations and implications, and the available preventive management of VC in patients with CKD.
Go to : 
BIOLOGICAL MECHANISM OF VASCULAR CALCIFICATION IN CHRONIC KIDNEY DISEASE
Difference between chronic kidney disease and general population
CKD patients have a higher prevalence of VC than the general population, which contributes to the high risk of cardiovascular morbidity and mortality.7) In CKD patients, the traditional factors alone do not fully explain the high prevalence of VC. Although the precise underlying mechanisms of VC in CKD are not fully understood, previous studies have suggested that the CKD patients have specific contributors to the development of VC.8)9) Emerging evidence indicates that non-traditional risk factors including uremic toxins, CKD-mineral and bone disease (CKD-MBD), oxidative stress, and inflammation may contribute to the development of VC and cardiovascular disease in CKD patients.4)10)
It is well known that VC is a highly controlled process mainly regulated by the vascular smooth muscle cells (VSMCs).11) In the physiological conditions, VSMCs have a contractile phenotype and maintain the structural and functional homeostasis of the vessel wall. However, under pathological conditions, VSMCs transform into osteoblast-like cells and secrete collagenous extracellular matrix.12) In vitro studies have reported that serum derived from CKD subjects could induce calcification in human aortic smooth muscle cells. Under uremic conditions, the expression of markers of a contractile VSMC phenotype is downregulated, while those of a synthetic phenotype is upregulated.13)14) In patients with CKD, the VSMCs change their phenotype and proceed to VC through cellular proliferation, migration, and apoptosis.15)16)17) Other studies suggested that several factors relevant to CKD including uremic toxins, inflammatory molecules are associated with a phenotype switch and physiological function of VSMCs.16)18)
Burden of oxidative stress and inflammation
Oxidative stress is known to increase in correlation with declining renal function and increased oxidative stress may contribute to cardiovascular events in CKD patients.19) Since the kidney is one of the main sources of antioxidant enzymes, an impairment of the renal function is associated with decreased levels of these enzymes, and CKD patients have an impairment of antioxidant systems.20)21) Yamada et al.22) demonstrated that oxidative stress induced by uremic conditions influences the development of VC in CKD using an animal model. They also reported that antioxidant treatment ameliorated phenotypic changes in VSMCs, providing evidence for the clinical application of antioxidants to prevent VC in CKD patients. Huang et al.23) observed an elevation of oxidative stress in CKD patients and demonstrated a significant association between oxidative stress and VC. Their findings provide evidence supporting the pivotal role of oxidative stress in the phenotypic change of VSMCs in early CKD.
Inflammation also plays a critical role in the development of VC in CKD patients by inducing VSMC osteochondrogenic conversion.24)25) Previous studies have shown both systemic and local inflammation related to VC in CKD patients. Uremic toxins promote the development of an inflammatory state, and elevated plasma levels of inflammatory markers are observed in CKD patients. The levels of inflammatory markers are significantly associated with the prevalence of VC and cardiovascular events in CKD patients.26)27)28)29) Benz et al.30) demonstrated that the vessels obtained from early CKD patients showed an upregulation of inflammatory molecules. Other researchers also reported a more pronounced macrophage infiltration in coronary artery lesions of CKD.31) Mounting evidence indicates that inflammation triggers changes in the VSMCs and promotes the VC process by releasing calcified extracellular vesicles. It has been proposed that inflammation may activate the endoplasmic reticulum stress pathway, which is associated with increasing inorganic phosphate uptake and causes VSMC phenotypic transformation and mineral accumulation.32)
Chronic kidney disease-mineral and bone disease
CKD-MBD is a dysregulation of mineral and bone metabolism caused by CKD, which includes biochemical abnormalities of calcium, phosphate, vitamin D, parathyroid hormone (PTH), bone disorders, and calcification.33) Increased serum levels of calcium and phosphate are considered as the main risk factors for the development of VC in patients with CKD.34) Hypercalcemia was suggested to engage in VC by increasing Ca × P product and enhancing sodium-dependent phosphate cotransporters.35) Hyperphosphatemia is a common finding in CKD patients, which is caused by the reduction of renal excretion. It is assumed that the effects of hyperphosphatemia during VC development are primarily associated with sodium-dependent phosphate cotransporters.36) Previous studies have reported that elevated phosphate levels induce the expression of osteoblastic markers which stimulate the dysregulation of VSMCs. Within the vessel wall, increased levels of calcium and phosphate synergistically induce phenotypic changes in VSMCs.37)
Active process, not passive deposition of mineral
VC in CKD was initially considered to be a passive process by which minerals were deposited on the vessel wall by the dysregulation of calcium and phosphorus.38) However, recent evidence indicates that VC is an active and highly controlled process that involves the dysregulation and phenotypic transformation of VSMCs.10) In CKD patients, an increase in the calcification promoter level and a simultaneous decrease in the inhibitor level are associated with a high prevalence of VC. We previously investigated 90 iliac arteries obtained from the dialysis recipients at the time of transplantation to identify the characteristics of calcification.39) VC was observed in 77.8% of the participants using microscopic examination. Fetuin-A, an active calcific inhibitor, and C-reactive protein (CRP) stain was positive in 60.0% and 97.8% of patients, respectively (Figure 1). These findings indicate that the arteries in CKD patients significantly experience active calcification and a high rate of inflammation. We also identified a high correlation between a positive CRP stain score and calcification score, which suggests that local inflammation is closely related to calcification in the vessels of CKD patients.
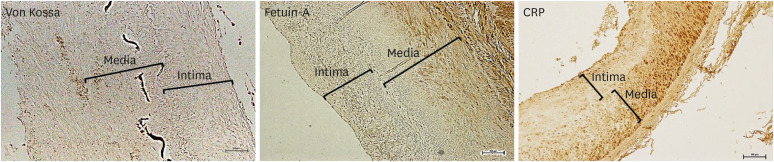 | Figure 1Immunohistochemistry of von Kossa, fetuin-A, and CRP in the iliac artery of kidney transplant recipients. Scale bar 100 μm. Original magnification×100.39)CRP = C-reactive protein.
|
Go to :
DISCOVERY OF MULTI-FACTORS INVOLVED IN VASCULAR CALCIFICATION
Fibroblast growth factor-23–Klotho axis
Fibroblast growth factor (FGF)-23–Klotho axis is a recently identified new player in the field of CKD-MBD and is one of the most notable substances in the pathogenesis of VC in CKD patients. FGF-23 is a bone-derived hormone that regulates phosphate and vitamin D homeostasis and the expression of klotho.40) An excess of FGF-23 and klotho deficiency are closely associated with VC in CKD.10)41) In animal studies, despite the induction of CKD, transgenic mice with klotho overexpression showed better renal function and lesser calcification compared with wild-type uremic mice.42) Krishnasamy et al. 43) reported that elevated serum FGF-23 levels are associated with the progression of arterial stiffness and aortic calcification in patients with advanced CKD. Other studies also demonstrated that an increased serum FGF-23 concentration was an independent predictor of VC in CKD patients, and suggested that FGF-23 is an independent biomarker of VC in CKD patients.41)44)45)46) Some studies have reported the protective effect of FGF-23. Zhu et al. 47) showed the expression profiles of FGF-23 during VC and demonstrated that FGF-23 prevents VC through the activation of the ERK1/2 pathway. They suggested that FGF-23 could be a potential therapeutic strategy. However, the treatment effect of FGF-23 on VC remains controversial and further studies are required to clarify this.
Osteoprotegerin
Osteoprotegerin (OPG) is a key regulator of bone remodeling and is involved in the vascular system. It acts as a decoy receptor binding to the receptor activator of nuclear factor-kappa B ligand and inhibits its interaction with the receptor activator of nuclear factor-kappa B.48) Recently, the interest in the role of OPG in the pathogenesis of VC has increased in the general population. OPG knockout mice showed early onset osteoporosis and extensive aortic medial calcification, suggesting the role of OPG as an inhibitor of VC.49) However, paradoxically, OPG expression was upregulated in VC lesions, and several clinical studies reported a positive correlation between OPG concentrations and the severity of VC.50) An increase in the OPG levels may be a result of compensatory response, for the protection of further vascular damage.51)52) Morena et al.53) showed that the serum levels of OPG were significantly associated with coronary artery calcification in the CKD patients and increased along with decreased renal function. Other studies with CKD patients also reported significant correlations between serum OPG concentrations and VC.54)55) Based on prior studies, OPG has been suggested as a potential biomarker of VC, and related studies are ongoing.
Fetuin-A
Fetuin-A is a circulating inhibitor of calcification and has anti-inflammatory properties thus, it exhibits a protective role against atherosclerosis.56) It binds early calcium-phosphate crystals and reduces the crystal growth and deposition.57) The levels of circulating fetuin-A showed an inverse relationship with CRP, a representative marker of inflammation.56) Mice deficient in fetuin-A develop prominent soft tissue calcification, indicating its role as a systemic inhibitor of ectopic calcification.58) In dialysis patients, the serum fetuin-A levels were decreased and negatively correlated with aortic and valvular calcification, calciphylaxis, and cardiovascular mortality.59)60) Recently, fetuin-mineral complex (FMC) has received attention in the pathogenesis of VC in CKD. The centrifugation of serum from rats with renal failure yielded a complex of fetuin-A and minerals; this complex apparently reflects the extra-osseous calcification stress.61) Hamano et al.62) investigated the clinical relevance of FMC in hemodialysis patients and suggested that the quantitative measurement of FMC, not supernatant or serum total fetuin-A, reflects extraosseous calcification stress. Further studies are required to validate these findings.
Go to :
DIAGNOSTIC IMAGING OF VASCULAR CALCIFICATION IN CHRONIC KIDNEY DISEASE
Comparison of multiple imaging modality
A variety of imaging modalities have been utilized in the diagnosis and monitoring of VC in CKD patients.63) Plain radiographs are mainly used to assess the calcification of large vessels. Atherosclerosis and calcification of the abdominal aorta can be evaluated using lateral abdominal radiography. Kauppila et al.64) suggested a simple scoring method to quantify VC in the abdominal aorta, which showed a significant correlation with arterial stiffness measured by the pulse wave velocity and coronary artery calcification measured by computed tomography (CT).65)66) The effectiveness of this scoring method has been validated not only in patients with mild to moderate renal dysfunction but also in patients with end-stage renal disease on dialysis.66) However, in our prior study using the iliac artery, no significant correlation was observed between radiologic and microscopic calcification. In addition, microscopic calcification was observed in 77.9% of patients without radiologic calcification. These findings raise the question of how sensitively plain radiographs can detect calcification.39) Despite these disadvantages, the information obtained from plain radiographs has the advantage that it is relatively easy to provide clinical relevance, considering that calcification observed on plain radiographs is significantly associated with cardiovascular events and mortality.67)68) Furthermore, plain radiography is widely used because it is inexpensive and easily available.
Ultrasound is suitable for evaluating large superficial vessels, such as the carotid and femoral arteries.69) Previous studies reported that calcified carotid plaques detected by ultrasound were significantly associated with the traditional risk factors of cardiovascular disease 70)71)72) and was an independent risk factor for cardiovascular events in CKD patients.73) Ultrasound has limitations that are operator dependent, subjective, and it is difficult to recognize the difference between intimal and medial calcifications. However, ultrasound could be a convenient alternative for VC evaluation because it can examine each vessel in detail without radiation exposure, detect the presence and extent of VC, and measure the pulse wave velocity, which is an index of arterial compliance and stiffness.63)69)74) Furthermore, in patients with CKD, ultrasound is often performed before dialysis vascular access creation. It provides information on the vascular status and calcification and assists preoperative planning and vascular mapping.75)76)
CT is more sensitive and objective for VC evaluation than other imaging modalities. Several studies have shown that the quantification of VC by CT has a high sensitivity and specificity and has a good prognostic value for cardiovascular events.63)77) The clinical significance of coronary artery and aortic calcification detected by CT is well established. Agatston et al.78) suggested a scoring system to quantify coronary artery calcification, which predicts cardiovascular events with high sensitivity and specificity. CT is considered as a reference standard for assessing VC because it accurately detects and quantifies VC. Furthermore, CT has the advantage of monitoring the progression of VC over a period of time and obtaining additional information such as valvular calcification and bone density.79) The limitations of CT lie in the high risk of radiation exposure, high cost, and low accessibility.80)81) In addition, because it is difficult to distinguish between intimal and medial calcification, its use for risk stratification is limited in CKD patients. Some studies have suggested reducing this limitation through a combination of biomarkers and other risk factors. Further studies are needed in this field.4)82)83)
Screening of vascular calcification
The Kidney Disease Improving Global Outcome (KDIGO) work group recommends screening for VC in CKD patients using either lateral abdominal radiography or CT.84) At the time of this review, there were several controversies regarding the benefit of screening for VC in CKD patients. Although the association between the VC and poor cardiovascular outcomes in CKD patients is well established, the impact of risk factor modifications through screening is not clear and there is no proper treatment for improving the clinical outcomes. Therefore, the KDIGO work group weakly recommends VC screening in patients with CKD.
Go to :
CLINICAL PRESENTATION OF VASCULAR CALCIFICATION IN CHRONIC KIDNEY DISEASE
Intimal vascular calcification
Intimal VC is closely associated with atherosclerotic plaque formation.85) Traditional cardiovascular risk factors, including older age, smoking, hypertension, diabetes, obesity and hyperlipidemia provokes the formation of atherosclerotic plaques, and intimal calcification occurs in the plaque as a late event in atherosclerosis (Figure 2A). CKD patients already have a number of traditional risk factors for cardiovascular disease, and atherosclerotic arterial lesions are more prevalent in CKD patients.86)87)88) In addition, the progressive decline of renal function leads to the further progression of atherosclerosis.89)90) Consequently, the burden of atherosclerosis is considerably increased in CKD patients, and excess calcium and phosphate precipitates facilitate the process of intimal calcification and a higher frequency of intimal calcification.91)92)
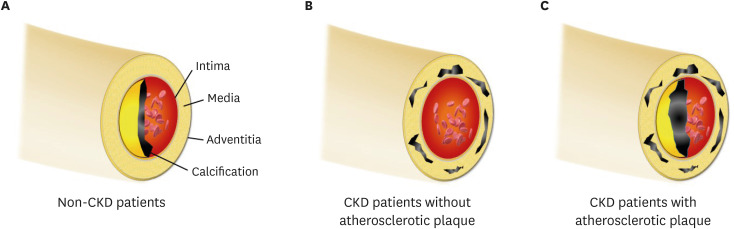 | Figure 2(A) In patients without renal dysfunction, calcification is deposited in the intimal layer; (B) CKD patients obtain the medial calcification in the absence of the atherosclerotic factor, because mineral bone disorder accelerates the calcific process; (C) In clinical practice, intimal and medial calcification frequently co-exists in CKD patients, because they already had several traditional risk factors. Notably, the intimal or medial calcific burden is greater in CKD patients than in non-CKD patients.CKD = chronic kidney disease.
|
Medial vascular calcification
Medial calcification theoretically appears to be more specific to CKD patients than intimal calcification, considering the pathogenic similarity between bone ossification and medial calcification.93)94) Hypercalcemia, hyperphosphatemia, secondary hyperparathyroidism and other CKD-MBD-related factors trigger mineral deposition on the medial layer of the vascular wall; also, the formation of micro-calcific foci accelerates the accumulation of additional calcification.18 Therefore, medial VC can frequently occur in CKD, despite the absence of traditional risk factors (Figure 2B).95) Arterial medial calcifications are not directly related to cholesterol deposits and do not induce occlusive lesions in the lumen. However, medial calcification is associated with an increased pulse pressure, arterial stiffness, poor vascular compliance and left ventricular hypertrophy.85)96) These changes lead to an increased rate of future cardiovascular events and mortality risk in the CKD population.97)
Which type of vascular calcification plays leading roles in chronic kidney disease?
The intimal and medial VCs represent different clinical entities with distinct pathogenic signatures and it would also appear that the basic pathogenic principles remain relevant in the CKD population (intimal calcification - atherosclerosis and medial calcification - loss of the cushioning function). However, the CKD patients retain common risk profiles and signaling pathways, which overlap in intimal and medial calcifications.30) In particular, oxidative and inflammatory signaling are the common pathways for both types of VC, and uremic conditions considerably activate these two pathways.98)99) Therefore, intimal and medial VC in CKD patients can frequently occur at the same time and several studies have reported the co-existence of the two types of VC (Figure 2C).100)
Go to :
CLINICAL IMPLICATION OF VASCULAR CALCIFICATION IN CHRONIC KIDNEY DISEASE
Different presentation of vascular calcification in different chronic kidney disease setting
Lower renal function increases the burden of VC, and its prevalence is substantially increased in CKD patients compared to patients without renal impairment.101)102)103) In non-dialysis-dependent CKD, VC is observed with a variable prevalence range around 30% to 60% patients.104) There is an inverse relationship between estimated glomerular filtration rate (eGFR) and VC score independent of traditional risk factors (Figure 3).105)106) In addition, CKD patients with a greater severity of coronary calcification score are expected to show a more rapid progression of calcification.107)108)
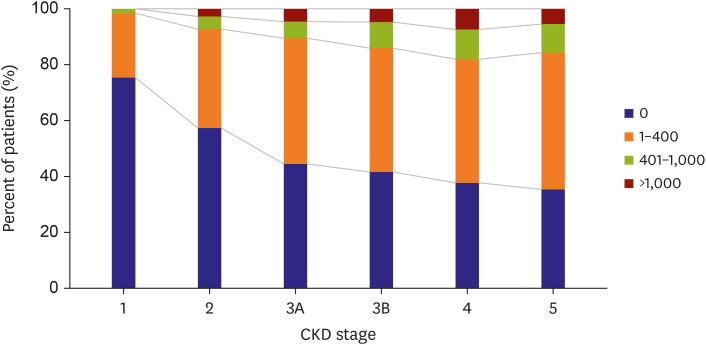
The prevalence and progression of VC increase dramatically, if CKD progression arrives at the latest stages. It has been reported that VC is detected in more than 50% of patients receiving dialysis treatment, and they have a 15–30% annual increase in coronary calcification.109)110)111) In our previous studies, it was also found that 40–45% of hemodialysis patients showed VC, which was detected in the aortic area using plain chest radiography.96)112)113) Notably, we found a strikingly increased prevalence of VC in the microscopic examination of the iliac artery compared to those on plain radiography of the aorta.39) The detected intimal calcification using von Kossa stain was observed in 62% of dialysis patients and medial calcification in 74% of the patients, while radiologic calcification was detected in 24% of the patients. These findings suggest that the actual prevalence of VC may be higher than previously reported.
Renal transplant recipients gain recovery of renal function from the successful engraftment of donor kidney. The restoration of the renal function allows for the elimination of multi-factors and toxins that play a role in promoting VCs. In addition, it is expected that CKD-MBD resolves spontaneously, because secondary hyperparathyroidism and impaired phosphate excretion disappear. Several studies have investigated whether VC progresses or regresses after successful renal transplantation and found that medial arterial calcification appeared to slow the progression. However, most renal transplant recipients take over the calcification trait from the dialysis days and these are not spontaneously regressed.114) Indeed, the transferred calcification from dialysis acts as strong predictor for the progression of calcification after transplantation and coronary arterial calcification progresses as approximately 10% increase/year after transplantation.115)116)117) In addition, secondary hyperparathyroidism continues in 40% of renal transplant recipients and persistent CKD-MBD is a significant determinant of progression of VC after transplantation.118)119)120) The current data support that all types of VC (intimal and medial) is gradually deteriorated after renal transplantation.
Cardiovascular consideration of vascular calcification
Cardiovascular events are the most common cause of death in CKD patients and VC is currently established as an uncontroversial risk marker of incident cardiovascular event.121)122)123) The presence of VC irrespective of the site is associated with a several-fold increase in the risk of cardiovascular morbidity and mortality in the entire spectrum of CKD patients and renal transplant recipients.124)125) The KDIGO international clinical practice guideline suggests that CKD patients with VC need to be considered as having the highest risk of incident cardiovascular events.126) In addition, VC is closely related to functional disorders of the cardiovascular system in CKD patients. Intradialytic hypotension is the most common hemodynamic dysfunction in hemodialysis patients and we found that VC is independently associated with a higher risk of intradialytic hypotension.113) The significant synergistic interaction between VC and intradialytic hypotension was also observed in the prediction of all-cause mortality and cardiovascular events, suggesting that the prognostic significance of VC is linked to the functional impairment in the hemodynamic system.
In the general population, the coronary arterial calcification score correlates well with the presence of atherosclerotic plaques. Nonetheless, the association between the severity of calcification and impaired perfusion is not strong in CKD patients.127) A recent report showed that the coronary artery calcification showed a weak relationship with myocardial ischemia, suggesting that coronary artery calcification may not be a good reflection of the atherosclerotic burden in CKD patients.128) As the calcification deposits in CKD patients are more active in the medial layer than those in patients with a normal renal function, the coupling power between the calcification score and ischemia becomes weaker. The anatomic pathology of atheromatous plaques provides additional constructive evidence. Previous studies on direct pathologic examination found that the volume of atheromatous plaque was not the main distinguishing factor of atherosclerosis between CKD and non-CKD patients. The differences originate in the composition of the plaque and calcific content (Figure 2A and C).31)129)130) CKD patients have more intense calcification on the intimal layer with additional medial calcification, which may not allow the VC score to fully reflect the atherosclerotic burden.
We reasonably question how reliable calcification is a predictor of coronary atherosclerotic events, not the overall cardiovascular mortality and survival rate. Undoubtedly, the dialysis patients with higher coronary artery calcification scores are significantly associated with coronary atheroma.131)132) In addition, higher calcification scores clearly predict the incidence of atherosclerotic coronary disease in CKD patients. However, the cutoff value of calcification score seems to be higher than non-CKD patients.133) Further studies are needed to determine the extent of calcification for the risk stratification of atherosclerotic events in CKD patients. The development of a new diagnostic modality to differentiate intimal and medial calcifications would be highly useful for atherosclerotic risk assessment of VC in CKD patients.
Non-cardiovascular consideration of vascular calcification
CKD patients are considerably exposed to calcification-prone conditions and any arterial system in our body presents calcific deposition. VC also evokes functional impairment in large and small arteries and causes problems in the non-cardiovascular system (Figure 4). We previously evaluated the relationship between VC and retinopathy, based on this plausible hypothesis.134) We found that retinopathy was significantly associated with the presence of aortic calcification, suggesting that large vessel calcification is linked with microvascular dysfunction. Furthermore, the combination of retinopathy and VC was stronger in predicting renal function decline than retinopathy alone. These findings support the concept that VC has prognostic implications in non-cardiovascular organs. The effect of VC on the renal outcomes was also associated with renal transplant recipients. Intimal calcification was significantly associated with a faster decline in the renal allograft function and a lower graft survival rate.135) The deposition of fetuin-A, a key regulator of calcific process, further reduces the allograft survival rate and is an independent predictor of renal graft function decline.
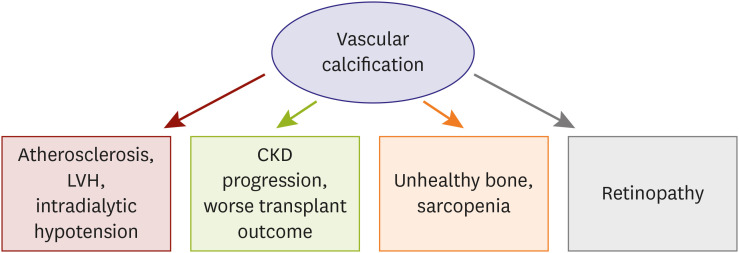
Secondary hyperparathyroidism and CKD-MBD are powerful inducers of VC in patients with CKD and it serves as a connecting link to bone health status. Several clinical studies have demonstrated that VC is associated with bone histomorphometry, low bone activity, low bone volume, and abnormal bone turnover in dialysis patients, suggesting an interplay between the bone health and VC.136)137)138) Notably, these studies consistently show that the mineralization in bone and vessel counterworks, while VC and bone mineralization share common pathophysiologic processes.139)140) CKD-MBD leads to bone resorption, decreased bone mineralization, and increased calcium efflux from bones promoting a positive calcium balance to vascular mineralization and calcification. This inverse relationship has been extended to recent research, which shows that VC is independently associated with incident fractures in dialysis patients.141)142)
It has been reported that increased adipose tissue volume and low muscle mass are positively associated with prevalent aortic and coronary calcification.143)144) Studies on CKD patients showed similar trend. Lee et al. 145) analyzed the nationwide prospective cohort of CKD in Korea and reported that normal body mass index with central obesity has increased risk of coronary artery calcification, and adipose tissue volume has a significant correlation with VC. 146)147) In addition, the prevalence of low muscle mass is increased as CKD is more advanced 148) and sarcopenia in hemodialysis patients shows an independent relationship with VC.149) These results suggest that pathogenesis of obesity, sarcopenia and VC may involve some shared mechanism, and inflammation is potential common pathway of them.
Go to :
MANAGEMENT OF VASCULAR CALCIFICATION IN CHRONIC KIDNEY DISEASE
Significance of chronic kidney disease-mineral and bone disease management in vascular calcification
VC is rarely observed to regress, and the primary goals of management are the prevention and stabilization of the existing calcifications. The optimal management of CKD-MBD is a fundamental key in determining the progression of VC, and the presence of an effective prevention method is the distinct differences in CKD compared to those of other metabolic conditions. The biochemical components of CKD-MBD are serum calcium, phosphate, and PTH levels, and nephrologists attempt to control all of these components using dialysis treatment and medications.150)151) The achievement of CKD-MBD parameters in the target range significantly reduced the risk of VC, cardiovascular mortality and all-cause death (Figure 5).92)
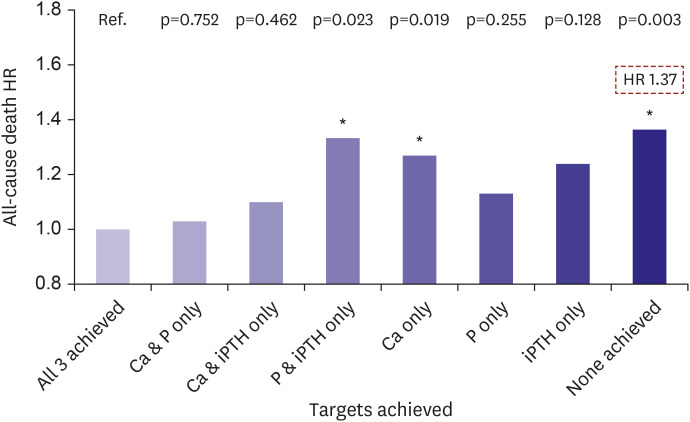 | Figure 5Target achievement of the CKD-MBD parameters and mortality risk in dialysis patients. KSN News Factsheet Vol. 18 (May 2020).Ca = calcium; CKD-MBD = chronic kidney disease-mineral and bone disease; HR = hazard ratio; KSN = Korean Society of Nephrology; P = phosphate; iPTH = intact parathyroid hormone.
|
PTH suppressing agents
Increased PTH levels are associated with deteriorated clinical outcomes in CKD patients, and treatment strategies focus primarily on the reduction of PTH levels. However, lower levels of PTH are also associated with an increased risk of mortality, and PTH should be controlled in the target range, which is different between CKD stages.126) Calcimimetics, paricalcitol, and active vitamin D are the three major agents for PTH reduction. Active vitamin D suppresses the proliferation of parathyroid cells; however, it also increases the absorption of calcium and phosphate from vitamin D receptors in the gastrointestinal tract. Therefore, the administration of vitamin D at therapeutic doses increased the serum calcium and phosphate levels, despite of PTH reduction. This phenomenon promotes the precipitation of calcium and phosphate products and enhances the progression of VC. The K/DOQI clinical practice guidelines recommend that serum calcium and phosphate should be monitored regularly with the use of active vitamin D compounds. Some experimental studies demonstrated that the therapeutic doses of vitamin D supplementation had protective effects against CKD-stimulated VC and low circulating levels of vitamin D was associated with greater calcification score.152)153) However, patients with higher level of vitamin D also revealed advanced VC and there is a lack of clinical studies on the bimodal association between vitamin D supplementation and VC, particularly in a prospective randomized design. Therefore, additional evidence is required to translate the bimodal effect of vitamin D into clinical practice.
Paricalcitol is a synthetic vitamin D analogue that works on the parathyroid gland more specifically than on non-parathyroid organs and showed greater activity in PTH reduction than other PTH suppressing agents.154) The higher selectivity of paricalcitol on the parathyroid gland reduces hypercalcemia and hyperphosphatemia. In addition, the dialysis patients receiving paricalcitol have a significant survival advantage compared to those receiving standard forms of active vitamin D compounds.155) Nevertheless, paricalcitol retains the mineral absorption effects on the gastrointestinal tract and is less effective in preventing the progression of VC.156) In contrast, calcimimetics act only on the calcium sensing receptor of the parathyroid gland and the serum calcium level is moderately decreased through PTH suppression. Therefore, of all the PTH suppressing agents, cinacalcet showed the best effectiveness in preventing VC and can reduce the risk of major adverse cardiovascular events and bone fracture.157)158)159)
Phosphate binder
CKD patients exhibit an impaired renal excretion of phosphate, and dialysis treatment does not fully eliminate the phosphate to the extent that it maintains normal the serum phosphate levels. Therefore, phosphate binders are usually administered to manage hyperphosphatemia in the advanced stages of CKD. The phosphate binders are classified into calcium-or non-calcium-containing regimens and these two types of phosphate binders show different results in the prevention of VC. Non-calcium-containing phosphate binders are more effective in avoiding a positive calcium balance in blood vessels; additionally, this benefit is translated into clinical practice. In dialysis patients, the non-calcium-containing phosphate binders interrupt the progression of VC and the protective effect against VC is more prominent in the patients with preexisting VC.160)161) In addition, there is some evidence that non-calcium-containing phosphate binders improved the survival rate in non-dialysis-dependent CKD and dialysis patients.162)163) Consequently, recent guidelines suggest that calcium-containing phosphate binders should be restricted and that non-calcium-containing regimens are preferred in the presence of VC.84)
Warfarin
Warfarin, a vitamin K antagonist, is used clinically as an oral anticoagulant for the management of atherothrombotic cardiovascular disorders. In CKD patients, the prevalence of atrial fibrillation is higher than in non-CKD patients, and the risk of major adverse cardiovascular outcomes is synergistically increased in the coexistence of CKD and atrial fibrillation.135) Therefore, the necessity of anticoagulation is increased in CKD patients. However, warfarin promotes calcification because it suppresses γ-carboxylation of the matrix Gla protein, an inhibitor of calcification. A number of studies implicate warfarin treatment in the development of VCs, and the high prevalence of vitamin K deficiency in CKD patients may potentiate the calcification-inducible effect of warfarin.164)165)166) Notably, the clinical benefit of warfarin is also disputed in CKD patients. It has been report that the incidence of stroke was even higher in dialysis patients on warfarin than in those on aspirin, clopidogrel, or no treatment.167) In non-dialysis-dependent CKD, warfarin treatment was observed to increase the risk of bleeding.168)
The adverse effects of warfarin on VC question whether the attractive options of new oral anticoagulants are valid in CKD patients to minimize VC and other adverse effects. In an experimental model of atherosclerosis, rivaroxaban showed a greater effect on the prevention of VC when compared to warfarin.169) Several human studies demonstrated that new oral anticoagulants lead to slow down the progression of calcification compared to warfarin, suggesting the benefit of new oral anticoagulants in clinical practice.170)171)172)173) In a recently published study on hemodialysis patients, switching from a vitamin K antagonist to rivaroxaban also revealed favorable effects in reducing the risk of life-threatening and major bleeding. However, rivaroxaban with vitamin K2 supplementation showed no significant protection against VC progression in hemodialysis patients.174) The authors suggest that VC already progressed in studied participants to the extent that intervention treatment is ineffective and that the use of rivaroxaban at earlier stages of CKD might be beneficial in preventing VC. Therefore, we need additional evidence to opt for the newly developed anticoagulants over warfarin in patients with CKD.
Go to :
CONCLUSION
In this review, we showed that VC in CKD is a highly organized process, and that uremic toxins, CKD-MBD, inflammation, and oxidative stress have contributed to the development of VC. Several factors involved in the pathogenesis of VC in CKD have been discovered and suggested as biomarkers, but further studies are needed. The clinical presentation of VC in patients with CKD also differs from that in the general population. The prevalence of VC and rate of VC progression change with different CKD status. The association between a higher VC score and greater burden of atherosclerosis is modified in CKD patients, and the prognostic significance of VC is linked to non-cardiovascular organs. In addition, the management of VC is more focused on CKD-MBD, not atherosclerosis, indicating that the clinical implications of VC are different. We anticipate that this updated review, ranging from the pathophysiology to clinical implications, will help to improve understanding VC in CKD patients.
Go to :
 XML Download
XML Download