

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Obesity is characterized by the disruption of T cell homeostasis, which may cause tissue and systemic inflammation [1]. Several studies have reported increased numbers of Th1 and Th17 cells in the adipose tissue and splenocytes of obese mice [2345]. Interferon (IFN)-γ, which is secreted by Th1 cells, is closely related to metabolic dysfunction and autoimmunity [67]. Winer et al. [5] reported that insulin resistance was induced by IFN-γ-producing Th1 cells in obese mice. The IFN-γ levels in serum and IFNG messenger RNA (mRNA) levels in peripheral blood mononuclear cells (PBMCs) were higher in lupus patients compared with healthy people [89]. In type 1 diabetes patients, PBMCs that were reacted with insulinoma associated-2 peptides, natural epitopes of a single islet autoantigen, exhibited extreme polarization to IFN-γ positive cells, and this might have been a CD4 T cell response because it disappeared when CD4 T cells were depleted [10]. Interleukin (IL)-17 is also associated with autoimmunity, as an excessive increase in Th17 cells in obese mice contributed to the development and maintenance of experimental autoimmune encephalomyelitis (EAE) and accelerated the progression of colitis [11]. Taken together, these findings show obesity-induced systemic metabolic dysfunction may be caused by the abnormal regulation of T cell homeostasis.
The dysregulation of vitamin D metabolism has been observed in obesity [1213]. T cells express the vitamin D receptor (VDR) and are known to be the direct and indirect targets of vitamin D [14]. Vitamin D has been reported to inhibit the differentiation of T cells into Th1 and Th17 cells and the production of IFN-γ, IL-17A, and IL-22 [1516]. Bruce et al. [17] reported that CD4 T cells from VDR-knockout mice showed increased development into Th17 cells compared with wild-type mice under in vitro Th17- and Treg-culture conditions, suggesting that vitamin D is critical in regulating the distribution of T cell subsets.
Autophagy, which is important for maintaining cell and energy homeostases, is an intracellular degradation system [18]. Autophagy regulates the stimulation, proliferation, differentiation, and function of T cells [19]. T cells have a basal level of autophagy, which is induced in response to T cell receptor and common-γ-chain cytokine receptor signals. The regulation of autophagy in obesity has been investigated in several organs, including the pancreas, liver, heart, and adipose tissue [202122], and many studies have reported an association between autophagy and vitamin D. Zhao et al. [23] found that the mRNA levels of LC3 in PBMC were lower in a severely vitamin-D-deficient group (serum 25(OH)D3 levels < 10 ng/mL) compared with a vitamin-D-insufficient group (serum 25(OH)D3 levels 10–30 ng/mL). According to Yuk et al. [24], 1,25(OH)2D3 treatment upregulated the mRNA levels of BECN1 and ATG5 in human monocytes and the fusion of autophagosomes and lysosomes in human monocytes and macrophages was also upregulated by 1,25(OH)2D3. These results imply that vitamin D affects autophagy in immune cells. However, a limited number of studies have investigated the effects of vitamin D on autophagy in T cells.
The aim of this study was to investigate the effects of in vitro vitamin D treatment on the function of T cells and the expression of proteins involved in autophagy in high-fat diet-induced obese mice. We evaluated whether in vitro 1,25(OH)2D3 treatment affects proliferative capacity, the expression of a surface marker and transcription factors, the production of cytokines related to T cell function, and the expression of autophagy-related proteins in the T cells of obese and control mice.
Go to : 
MATERIALS AND METHODS
Animals and diets
Five-week-old male C57BL/6 mice (Central Laboratory Animal, Inc., Seoul, Korea) were housed in a specific pathogen-free room with an environmentally controlled temperature (23°C ± 1°C), relative humidity (50% ± 10%), and light/dark cycle (12/12-hour light/dark). After a 5-day adaptation period, the mice were randomized into two groups and fed experimental diets differing in fat content for 12 weeks. The CON group was fed the control diet (10% kcal fat, D12450B, Research Diets, Inc., New Brunswick, NJ, USA) and the HFD group was fed a high-fat diet (45% kcal fat, D12451, Research Diets, Inc.). The experimental diets and drinking water were provided ad libitum. Food intake was recorded four times per week, and body weight was measured once a week. At the end of the experimental period, the mice were fasted for 12 hours and euthanized by CO2 asphyxiation. All experimental procedures were approved by the Institutional Animal Care and Use Committee of Seoul National University (approval No. SNU-181029-6).
Preparation of 1,25(OH)2D3 solution
A stock solution of 10 μM 1,25(OH)2D3 (Sigma Aldrich, St. Louis, MO, USA) was prepared in 99.9% ethanol and filtered through a 0.22-μm filter. Further dilution was performed by diluting with complete Roswell Park Memorial Institute (RPMI) medium containing 10% fetal bovine serum (FBS; GibcoBRL, Grand Island, New York, USA) for cell culture. The complete RPMI was prepared with the addition of 100,000 U/L penicillin (GibcoBRL), 100 mg/L streptomycin (GibcoBRL), 25 mmol/L HEPES (Sigma Aldrich), and 2 mmol/L L-glutamine (GibcoBRL) in RPMI 1640 medium (Lonza, Walkersville, MD, USA).
Isolation of T cells from spleen
Spleens were removed and put into sterile complete RPMI. A single-cell suspension was prepared by homogenizing each spleen with sterile frosted glass slides. Splenocytes were centrifuged at 700 rpm for 25 s at 25°C to remove tissue debris, then resuspended in complete RPMI. Red blood cells were lysed using Gey's solution. Cells were washed twice and suspended in complete RPMI with 10% FBS. Cell viability was determined by the trypan blue exclusion test. To purify T cells, the cell suspension was incubated with a mixture of mAbs (Pan T cell Isolation Kit II or CD4+ T cell Isolation Kit; Miltenyi Biotec, Bergisch Gladbach, Germany) and negatively selected using the magnetic-activated cell sorter QuadroMACS separator and LS column (Miltenyi Biotec) according to the manufacturers' instructions.
In vitro 1,25(OH)2D3 treatment
Purified T cells were cultured in a 24-well plate (1 × 106 cells/well) for 72 h with complete RPMI containing 10% FBS. T cells from the same animal were cultured with either 10 nM 1,25(OH)2D3 solution or 0.1% ethanol (vehicle control) and with or without stimulation with 5 μg/mL plate-bound anti-CD3 (clone 145-2C11; BD Biosciences, San Jose, CA, USA) and 2 μg/mL soluble anti-CD28 (clone 37.51; BD Biosciences) antibodies during the entire culture period. Cells were incubated at 37°C in 5% CO2. After 72 h, the supernatant was collected for enzyme-linked immunosorbent assay (ELISA) analysis, and T cells were harvested for western blot analysis. CD4+ T cells were used for flow cytometric analysis. The experimental design and cell culture process are depicted in Fig. 1.
T cell proliferation
Purified T cells were plated into a 96-well round-bottom cell culture plate (2 × 105 cells/well; Thermo Fisher Scientific, Waltham, MA, USA), cultured with or without 5 μg/mL plate-bound anti-CD3 mAb (clone 145-2C11; BD Biosciences) and 2 μg/mL soluble anti-CD28 mAb (clone 37.51; BD Biosciences), and treated with 10 nM 1,25(OH)2D3 or a vehicle control for 72 h. Cells were pulsed with 0.5 uCi of [3H] TdR (PerkinElmer, Waltham, MA, USA) in 20 μL of complete RPMI for the last 8 h. The cells were harvested on filter paper using a MicroBeta FilterMate-96 Harvester (PerkinElmer), and the proliferation was quantified by measuring the amount of [3H] TdR incorporated into the DNA, as determined by the MicroBeta2 Plate Counter (PerkinElmer). Data are expressed as counts per minute.
Flow cytometric analysis
For the FACS analysis, 1 × 105 CD4+ T cells were resuspended in a FACS-staining buffer (0.09% sodium azide, 1% FBS, 1 × PBS based, 0.22-μm filter-sterilized) and stained with fluorescence-labeled antibodies specific for CD25-conjugated FITC (clone 7D4; BD Biosciences) or the isotype control at 4°C for 30 min. Then, cells were incubated with Fixation/Perm working buffer for intracellular staining (Foxp3/Transcription Factor Staining Buffer Set; eBioscience, Inc., San Diego, CA, USA). After incubation, the cells were resuspended in the perm diluent and reacted with the antibodies RORγt-conjugated PerCP-cy™5.5 (clone Q31-378; BD Biosciences) and Foxp3-conjugated Alexa Fluor 488 (clone MF-14; Biolegend, San Diego, CA, USA) or the isotype controls. After staining, cells were washed and resuspended in a cell fixer (4% formaldehyde, 1 × PBS based), and analyzed using FACS Calibur II and FlowJo software version 10.6.2. (BD Biosciences).
Quantification of cytokine production
Supernatant was collected after 72 h of cell culture. The levels of IFN-γ, IL-17A, IL-4, and IL-10 produced by T cells were determined using Mouse ELISA IL-17A (Invitrogen, Carlsbad, CA, USA), IFN-γ, IL-4, and IL-10 kits (BD Biosciences) according to the manufacturers' instructions. The absorbance was measured at 450 nm with a microplate spectrophotometer (Spectramax iD3; Molecular Devices, San Jose, CA, USA).
Western blot analysis
Protein was extracted from T cells using radio-immunoprecipitation assay buffer (50 mM Tris-HCl [pH 7.4], 1% NP-40, 0.25% Na-deoxycholate, 150 mM NaCl, 1 mM EDTA, 1 mM PMSF, 1 mM Na3VO4, 1 mM NaF, 1 mM Na-pyrophosphate, 1 mM β-glycerophosphate, 10% glycerol, protease inhibitor cocktail tablet [Roche, Basel, Switzerland]). Protein lysates (30 μg) were electrophoresed on 13% SDS-PAGE gels and transferred to a polyvinylidene difluoride membrane. The membrane was incubated with the following antibodies: LC3A/B (1:1,000), SQSTM1/P62 (1:1,000), BECLIN-1 (1:1,000), and ATG12 (D88H11) (1:1,000), followed by HRP-conjugated anti-rabbit IgG (1:3,000). All antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). Specific bands on the membrane were visualized with chemiluminescence luminol reagent (Santa Cruz Biotechnology, Dallas, TX, USA) and developed using the JP-33 automatic X-ray film processor (JPI Healthcare, Seoul, Korea). The target bands were visualized and analyzed using Quantity One software (Bio-Rad, Hercules, CA, USA).
Statistical analyses
Statistical analyses were conducted using SPSS statistics software version 25 (IBM SPSS Statistics, Chicago, MI, USA). The student's t-test was used to determine the effect of the dietary fat amount. A paired t-test was performed to determine the effect of in vitro 1,25(OH)2D3 treatment. If the data did not follow normality, nonparametric tests, i.e., Mann-Whitney U test or Wilcoxon signed-rank test, were performed. All data are presented as mean ± SE and statistical significance was set at P < 0.05.
Go to :
RESULTS
Body weight, weight change, WAT weight, and food intake
There was no significant difference in the body weights at 0 weeks between the CON and HFD groups. After 12 weeks of feeding, the body weight of the HFD group was 30.7% higher than the CON group (P < 0.001). Weight gain (P < 0.001) and white adipose tissue weight (P < 0.001) were significantly higher in the HFD group than the CON group. The average food intake was significantly lower and average energy intake was significantly higher in the HFD group (both P < 0.001) compared with the CON group (Table 1).
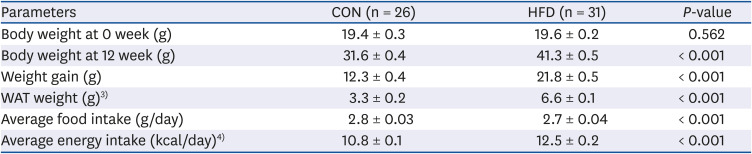
Table 1
Body weight, weight gain, adipose tissue weight, and food intake of mice in the CON and HFD groups1),2)
CON, control; HFD, high-fat diet.
1)The data are presented as mean ± SE.
2)Student's t-test was used to determine the effect of HFD. If the data does not follow the normality, Mann-Whitney test was performed.
3)WAT includes perirenal, intraperitoneal, epididymal, and subcutaneous fat.
4)Calculated from the average food intake and the energy density of the CON diet (3.82 kcal/g) and HFD (4.7 kcal/g).
![]()
T cell proliferation
The proliferative capacity of T cells from the HFD group tended to be lower than that of T cells from the CON group (39.5% lower, P = 0.069). In vitro 1,25(OH)2D3 treatment had no significant effect on T cell proliferation in both the CON and HFD groups (Fig. 2).
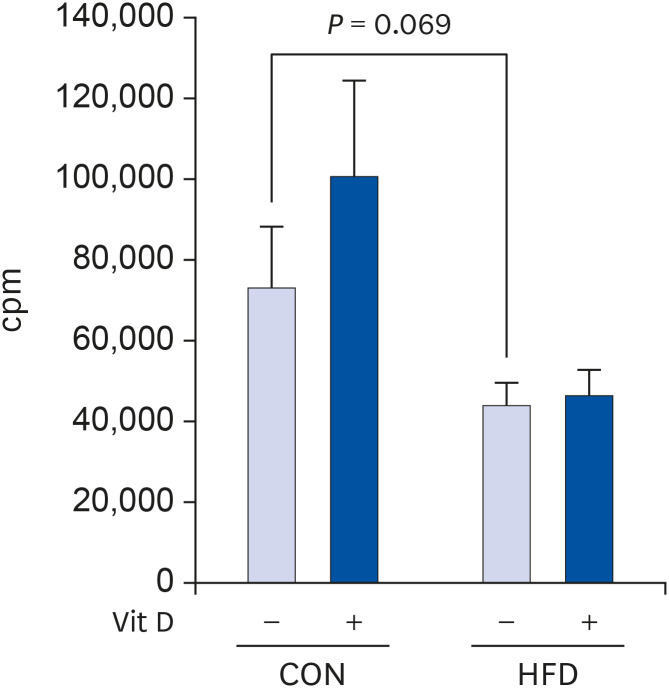 | Fig. 2Effects of HFD and in vitro 1,25(OH)2D3 treatment on T cell proliferation. Total T cells purified from splenocytes of CON and HFD groups were stimulated with 5 μg/mL plate-bound anti-CD3 mAb and 2 μg/mL soluble anti-CD28 mAb and treated with 10 nM 1,25(OH)2D3 or vehicle CON for 72 h. Cells were pulsed with 0.5 μCi of [3H] TdR for the last 8 h of culture. [3H] TdR uptake was determined by liquid scintillation counting. The data are presented as mean ± SE (n = 6 to 8 per group). A student's t-test and a paired t-test were performed to determine the effects of dietary fat amount and in vitro 1,25(OH)2D3 treatment.Vit D, vitamin D; CON, control; HFD, high-fat diet; mAb, monoclonal antibody.
|
Expression of surface marker and transcription factors of CD4+ T cells
The proportions of Th17 (RORγt+) cells and Treg (CD25+ Foxp3+) cells were unaffected by either fat amount or in vitro 1,25(OH)2D3 treatment (Fig. 3).
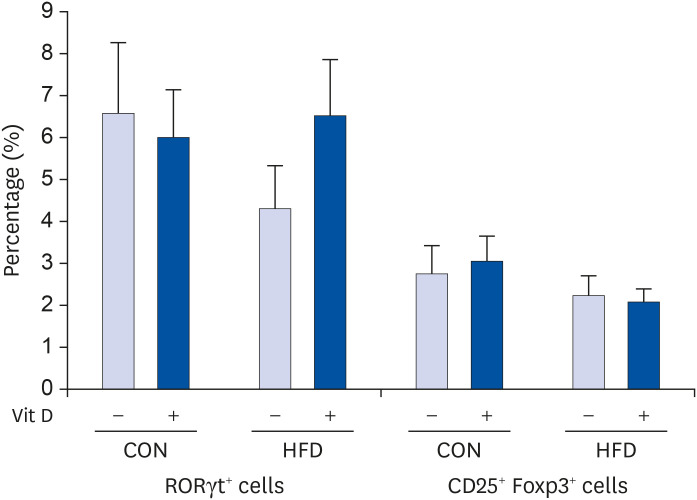 | Fig. 3Effects of HFD and in vitro 1,25(OH)2D3 treatment on the expression of surface marker and transcription factors related to Th17 and Treg in CD4+ T cells. CD4+ T cells purified from splenocytes of CON and HFD groups were incubated with 1,25(OH)2D3 (10 nM) or vehicle CON and stimulated with plate-bound anti-CD3 mAb and soluble anti-CD28 mAb for 72 h. Cells were harvested and analyzed by flow cytometry. Intracellular expression of RORγt was used as the marker related to Th17 cell. Intracellular expression of Foxp3 among cells positive for surface CD25 was used for identification of Treg cells. Values are presented as mean ± SE (n = 4 to 10 per group). Student t-test and paired t-test was used to determine the effects of dietary fat intake and in vitro 1,25(OH)2D3 treatment.CON, control; HFD, high-fat diet; mAb, monoclonal antibody.
|
Production of cytokines by T cells
The production of IFN-γ by T cells was significantly higher in the HFD group compared with the CON group (2.6-fold higher, P < 0.05) (Fig. 4A). However, in vitro 1,25(OH)2D3 treatment had no significant effect on IFN-γ production by T cells. IL-17A levels were not significantly different between the CON and HFD groups; however, in vitro treatment with 10 nM 1,25(OH)2D3 significantly decreased IL-17A levels in both the CON (34.8% less, P < 0.001) and HFD (41.6% less, P < 0.001) groups compared with the respective vehicle controls (Fig. 4B). IL-4 and IL-10 production was not affected by either fat amount or in vitro 1,25(OH)2D3 treatment (Fig. 4C and D).
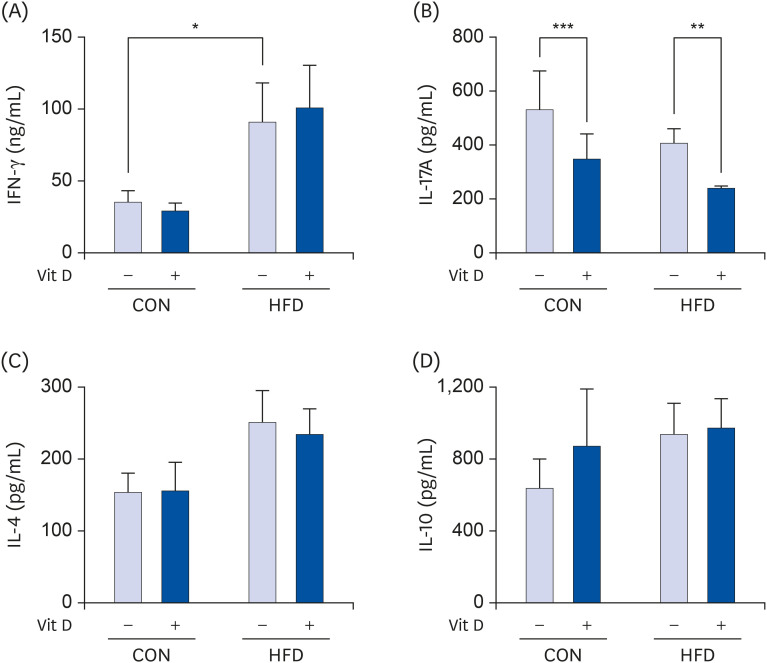 | Fig. 4Effects of HFD and in vitro 1,25(OH)2D3 treatment on production of cytokines by T cells. Total T cells purified from splenocytes of CON and HFD groups were cultured with 10 nM 1,25(OH)2D3 or vehicle (0.1% ethanol) and activated with anti-CD3 mAb (5 μg/mL) and anti-CD28 mAb (2 μg/mL) for 72 h. Supernatants were collected and assayed to determine the production of (A) IFN-γ, (B) IL-4, (C) IL-17A, and (D) IL-10 using ELISA. Data are presented as mean ± SE (n = 15 to 17 per group). A paired t-test was performed to confirm the effects of in vitro 1,25(OH)2D3 treatment. A student's t-test was used to determine the significant effects of dietary fat intake.IFN, interferon; IL, interleukin; CON, control; HFD, high-fat diet; mAb, monoclonal antibody.
*P < 0.05, **P < 0.01, ***P < 0.001.
|
Expression of proteins involved in autophagy in T cells
The LC3 II/I ratio was significantly higher in the HFD group compared with the CON group (57.4% higher, P < 0.05). In vitro treatment with 10 nM 1,25(OH)2D3 did not significantly affect the LC3 II/I ratio in both CON and HFD groups. The expression levels of SQSTM1/P62, BECLIN-1, and ATG12 were not significantly different between the CON and HFD groups, and in vitro treatment with 10 nM 1,25(OH)2D3 did not have a significant effect on these proteins (Fig. 5).
 | Fig. 5Effects of HFD and in vitro 1,25(OH)2D3 treatment on expression of proteins associated with autophagy in total T cells (A) Immunoblotting for LC3, ATG12, BECLIN-1, SQSTM1/P62, and β-ACTIN, and (B) densitometric analysis of protein expression. Total T cells purified from splenocytes of CON and HFD groups were incubated with 10 nM 1,25(OH)2D3 or vehicle CON, and stimulated with anti-CD3 mAb (5 μg/mL) and anti-CD28 mAb (2 μg/mL) for 72 h. Cells were harvested, and analyzed on western blot. The intensity of LC3, ATG12, BECLIN-1, SQSTM1/P62 in T cells was densitometrically measured and normalized with β-ACTIN. Data are presented as mean ± SE (n = 5 per group). Student t-test and paired t-test was used to determine the effects of HFD and in vitro 1,25(OH)2D3 treatment.CON, control; HFD, high-fat diet; mAb, monoclonal antibody.
*P < 0.05.
|
Go to :
DISCUSSION
In this study, we investigated the effects of in vitro vitamin D treatment on the proliferation, differentiation, and autophagy of splenic T cells in obese and control mice. T cell proliferation tended to decrease with obesity; however, the production of IFN-γ was higher in the obese mice compared with the lean controls. This increased IFN-γ production seemed to be partially related to the higher LC3 II/I ratio in the obese mice.
Our results show that the proliferation of T cells tended to be lower in the HFD group (39.5% lower). Several previous studies have reported lower proliferation levels of splenocytes and T cells from obese mice compared with those from control mice, which implies that obesity induces the dysfunction of immunity [252627]. Conflicting results have been observed for the effect of vitamin D on the proliferation of T cells. Lacey et al. [28] reported that when D10.G4.1 cells—the Th2 cell line of murine lymph nodes—were treated with 10 nM of 1,25(OH)2D3, they showed higher proliferative capacity than cells cultured without 1,25(OH)2D3. Whereas, in a study by Rigby et al. [29], the proliferation of phytohemagglutinin-stimulated PBMC was suppressed with 0.001–100 nM of 1,25(OH)2D3, suggesting vitamin D treatment has an inhibitory effect on T cell proliferation. However, as vitamin D also affects the maturation and function of unstimulated antigen-presenting cells (APC) [30], the inhibition of PBMC by 1,25(OH)2D3 might be an indirect consequence of its effect on the other immune cells in the culture. In this study, we showed that vitamin D does not directly affect the proliferation of T cells without the presence of APCs.
Our results show that the production of IFN-γ by T cells was higher in the obese mice compared with the controls. Consistent with our results, the proportion of IFN-γ+ cells in splenic CD4+ T cells stimulated with anti-CD3/anti-CD28 and the IFN-γ levels produced by PHA-stimulated splenocytes were reported to be higher in obese mice than control mice [24]. IFN-γ, which is secreted by CD4+ Th1 cells, CD8+ cytotoxic T cells, and various other immune cells, promotes macrophage activation, enhances antigen presentation, and regulates T cell subset differentiation [3132]. IFN-γ priming induces post-transcriptional changes that cause macrophage activation and inflammatory responses triggered by Toll-like receptor ligands [33]. Furthermore, excess IFN-γ in the culture medium leads to the impaired development of memory CD8+ T cells during infection, and increased protein expression of IFN-γ may contribute to insulin resistance [534]. According to a report by Vandanmagsar et al. [35], higher levels of Nlrp3 mRNA in the adipose tissue of obese mice induced the mRNA and protein expression of Th1 cytokine IFN-γ, which increased the activation of M1 macrophages and caused both insulin resistance and inflammation. This study confirmed there is an increased production of IFN-γ, specifically by T cells, in obesity. This could be one of the contributing factors aggravating the macrophage activation and inflammation often observed with obesity.
In vitro treatment with 1,25(OH)2D3 decreased the IL-17 production by T cells from both control and obese mice, but there was no difference in levels of IL-17 produced between control and obese mice. Obesity has been reported to promote an expansion of the Th17 T cell sublineage, and this has been suggested as the reason for more pronounced autoimmune disease associated with obesity [11]. However, it seemed that divergence exists among tissues with obesity regarding Th17 cytokine as IL-17 was increased in the liver while decreased in the ileum, colon, and white adipose tissue [36]. T cells purified from spleens of obese mice did not exhibit difference in IL-17 production compared with the lean mice in this study. In vitro treatment with 1,25(OH)2D3 inhibited differentiation of Th17 cells and oral treatment with 1,25(OH)2D3 in animal experimental model of EAE inhibited the onset of EAE and IL-17 positive cells in the splenocytes [37]. In our study, IL-17 production by purified T cells was inhibited by vitamin D in both control and obese mice. The significance of this needs to be investigated in the future study.
The ratio of LC3-II to LC3-I was higher but p62 expression was comparable in obese mice compared with the lean controls in our study. The increase in LC3 indicates there was increased autophagosome formation; however, the similar p62 levels in the two groups suggests that initiation of autophagy was inhibited because of reduced availability of autophagy precursors [3839]. Impaired autophagy reduces the clearance of apoptotic cells, resulting in tissue inflammation and the dysregulation of T cell homeostasis [40]. While IFN-γ induces autophagy by inducing LC3 puncta and LC3-positive phagosomes to eliminate intracellular antigens in macrophages [404142]. Considering the involvement of IFN-γ in macrophage autophagy, higher IFN-γ production by T cells in the obese mice in this study might be related to the higher LC3 II/I ratio. Although the mechanisms by which IFN-γ induces autophagy in macrophages have not been completely revealed, the p38 MAPK signaling pathway and the pathway involving immunity-related GTPase family M member 1 have been proposed to be the autophagy activation pathways induced by IFN-γ [4344]. Rincon et al. [45] showed that the production of IFN-γ by Th1 cells was significantly reduced when ConA-stimulated CD4+ T cells were treated with a p38 MAPK inhibitor, whereas IFN-γ production by Th1 cells was increased in constitutively activated MKK6 (upstream activator of p38 MAPK) transgenic mice. From these results, it appears autophagy in the T cells of obese mice may be activated through the pathway that activates autophagy in macrophages, involving the increased production of Th1 cytokines.
Vitamin D has been shown to regulate autophagy in many tissues and cancer cells and to accelerate anti-mycobacterial activity by increasing the mRNA levels of BECN1 and ATG5 and the fusion of autophagosomes and lysosomes in human monocytes and macrophages [2446]. Although autophagy has been shown to affect the regulation of T cell metabolism, studies investigating the effects of vitamin D on autophagy in T cells have been limited. The decreased expression of LC3 in PBMC in vitamin D-deficient patients suggests that vitamin D is important for autophagy in T cells [23]. Klug-Micu et al. [47] reported that soluble CD40-ligand-activation of human monocytes cultured in vitamin D-sufficient human serum upregulated the expression of CYP27B1 and VDR, and increased the proportion of LC3-II+ cells. Autophagy in monocytes was enhanced when CD3-activated T-cell clones were co-incubated with primary human monocytes in 10% vitamin D-sufficient human serum, suggesting that the T cell-mediated pathway might be associated with the vitamin D-dependent antimicrobial mechanism in human monocytes. However, we observed no significant effect of 1,25(OH)2D3 treatment on autophagy in T cells in this study.
In this study, only male mice were used. Sex differences in HFD induced obesity and metabolic syndrome as well as the T cell population have been reported [48]. Male mice developed adipose tissue inflammation, glucose intolerance, hyperinsulinemia with HFD feeding, while female mice were protected against-HFD-induced metabolic changes and maintained an anti-inflammatory environment in the intra-abdominal adipose tissue with expanded Treg cell population. We anticipated that vitamin D's effect on T cell function would be more evident in male mice which are more vulnerable to the HFD-induced changes. Non the less, effects of vitamin D on T cell function need to be determined in female mice in the future study.
In conclusion, our findings suggest that HFD-induced obesity affects the proliferation, cytokine production, and autophagy of T cells. Proliferation tended to decrease, while the production of IFN-γ and the LC3 II/I ratio increased, with HFD-induced obesity. The elevated Th1 cytokine production in obese mice compared with control mice may have contributed to increased levels of LC3. These results suggest that diet-induced obesity impairs T cell function and inhibits autophagy, resulting in the dysregulation of T cell homeostasis, which may contribute to the exacerbation of inflammation commonly observed with obesity. Vitamin D treatment did not affect autophagy of T cells, but inhibited the production of IL-17.
Go to :
 XML Download
XML Download