

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Tissues that are very active in energy metabolism are more likely to face metabolic challenges from the bioenergetic substrate and are susceptible to mitochondrial diseases [1]. Several genes from both the nucleus and mitochondria are required for mitochondrial formation and maturity. Thus, intercommunication between the mitochondria and nucleus is critical for highly metabolic tissues such as skeletal muscle, heart, liver, and adipose tissue.
The mitochondria are heavily involved in diabetes, insulin resistance, complications derived from diabetes [2], and cellular calcium (Ca2+) flux, an important process for mitochondrial function and metabolic diseases since Ca2+ signaling regulates various events related to cell death and energy metabolism [3-7]. Mitochondrial metabolic dysfunctions induced by insulin resistance are linked to abnormal Ca2+ flux and high reactive oxygen species (ROS) levels. The metabolic functions that regulate Ca2+ flux are also associated with glucose uptake and metabolism [4]. The nucleus responds to Ca2+ signals, yet the endoplasmic reticulum (ER) dynamically interacts with the mitochondria and regulates Ca2+ signaling. This process is essential for several metabolic processes and physiological functions [8], and is important for regulating normal mitochondrial biogenesis [5,9] and glucose metabolism [4].
The nucleus encodes the mitochondrial transcription factor A (TFAM), which activates transcription in the mitochondria and transmits a signal to the nucleus based on the mitochondrial state by regulating Ca2+ levels. It has been shown that it also prevents high-fat diet-induced insulin resistance [10]. Tfam-mutant mice developed diabetes and exhibited mitochondrial deoxyribonucleic acid (mtDNA) depletion, deficient oxidative phosphorylation, and abnormal mitochondrial structure [9]. This suggests that TFAM is involved in cellular metabolic function, possibly involving Ca2+ signaling.
This review focuses on how mitochondrial TFAM regulates Ca2+ signaling and how dysregulation of Ca2+ by the mitochondria leads to metabolic disease in type 2 diabetes mellitus (T2DM).
MITOCHONDRIA AND TFAM
Mitochondria play a key role in cellular physiology, and are responsible for producing cellular energy and essential metabolites, along with regulating apoptosis [11]. These functions are dependent on gene expression in both the mitochondria and nucleus and are regulated by communication between mitochondria and other organelles.
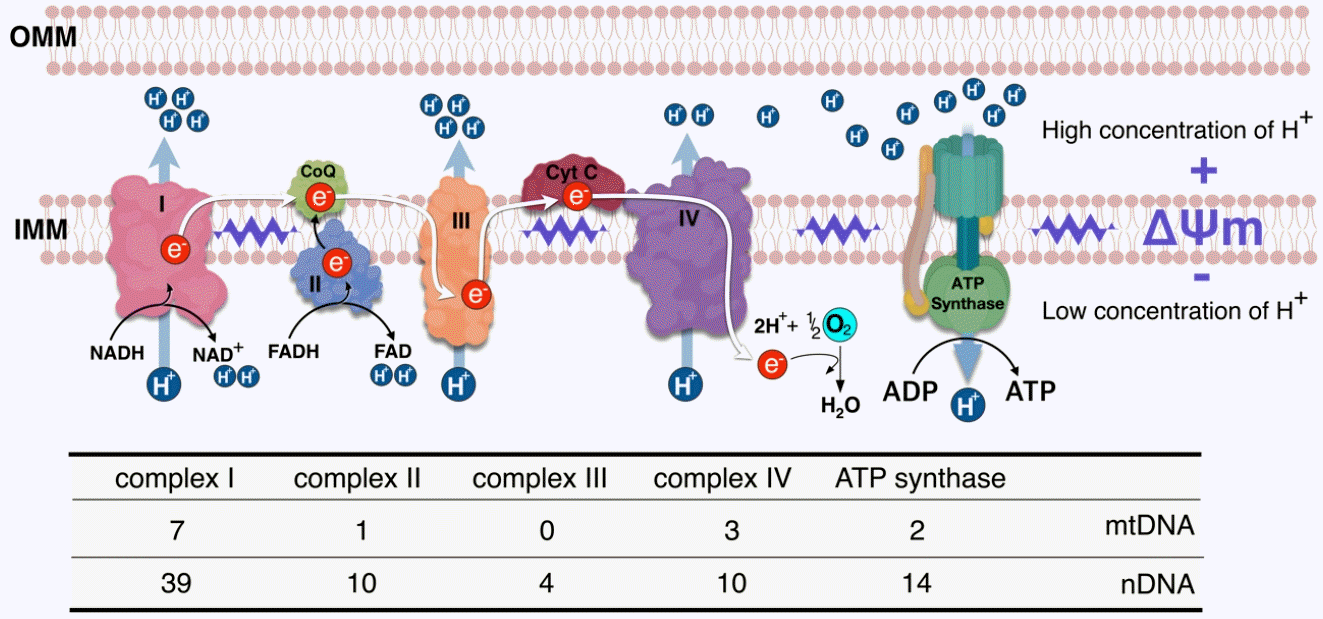
Biosynthesis of each electron transport chain (ETC) complex in the mitochondria is jointly regulated by the nucleus and mtDNA. Since several genes are required for mitochondrial biogenesis (Fig. 1), TFAM is an important regulator between the mitochondria and nucleus, since Tfam is expressed from the nucleus but acts on the mitochondria. TFAM regulates transcription of the 13 genes for ETC protein, 22 for transfer RNAs, and two for ribosomal RNAs encoded by mtDNA [12,13]. TFAM binds to mtDNA and enhances transcription in association with mitochondrial RNA polymerase and either mitochondrial transcription factor B1 (TFB1M) or B2 (TFB2M) [14]. In addition to binding to the specific region of the promoter, TFAM binds nonspecifically to random sites on mtDNA [15,16] which enhances the stabilization and maintenance of the mitochondrial chromosome [15-17] as well as regulation of mtDNA copy number [18]. Interestingly, since TFAM overexpression has been shown to increase mtDNA without alteration of respiratory capacity and mitochondrial biogenesis [10], it seems that the mechanisms underlying mitochondrial biogenesis and maintenance of mtDNA by TFAM are separately regulated. Moreover, TFAM overexpression increases the nuclear expression of various factors associated with Ca2+ signaling and glucose metabolism in mouse skeletal muscle [10]. TFAM is also involved in mitochondrial function including lower ROS via enhancing antioxidants, 5’ adenosine monophosphate-activated protein kinase (AMPK) activation, and mitochondrial uncoupling as well as regulation of membrane potential [10]. Therefore, intercommunication between the mitochondria and nucleus is closely regulated to cope with a sudden change in cellular energy challenges, with TFAM possibly playing an important role in this interaction.
NUCLEUS DISPATCHES TFAM TO THE MITOCHONDRIA
Signaling from the nucleus can regulate mitochondrial gene expression or mtDNA replication in response to cellular metabolic challenges or environmental signals [19], via TFAM. Nuclear respiratory factor 1 (NRF-1) can bind specific promoters of various nuclear genes required for mitochondrial respiratory function [20], which coordinates the expression of respiratory subunits with the mitochondrial transcriptional system [13]. NRF-1 also binds and activates the Tfam promoters [21], thereby inducing a cellular signaling cascade in skeletal muscle in response to exercise training [22,23]. In this context, Ca2+ treatment in muscle cell increases TFAM and NRF-1 protein levels as well as mitochondrial biogenesis [5,9], suggesting Ca2+ as a signaling molecule between cellular organelles. Redox reactions are activated in the mitochondria during exercise-induced oxidative phosphorylation and the incidence of NRF-1 binding to the TFAM promoter is increased under pro-oxidant conditions. However, Tfam expression is inhibited by deactivated NRF-1 [24], suggesting that Tfam expression mediated by NRF-1 is regulated under activated redox conditions. Interestingly, mtDNA depletion induces an increase in both along with oxidative stress [25]. NRF-1 and Tfam mRNAs are also increased in response to mitochondrial lipopolysaccharide-induced oxidative damage [26], suggesting that TFAM is increased to mitigate these cellular conditions, which, along with NRF-1, have been shown to decrease ROS via enhanced antioxidant and mitochondrial uncoupling [10].
Taken together, when the cellular environment undergoes metabolic challenge, including exercise or metabolic alteration, NRF-1 and TFAM are upregulated in the nucleus followed by TFAM translocation into the mitochondria to mitigate this change by regulating mitochondrial oxidative phosphorylation and redox valence.
Intercommunication between mitochondria and ER for intracellular Ca2+ flux
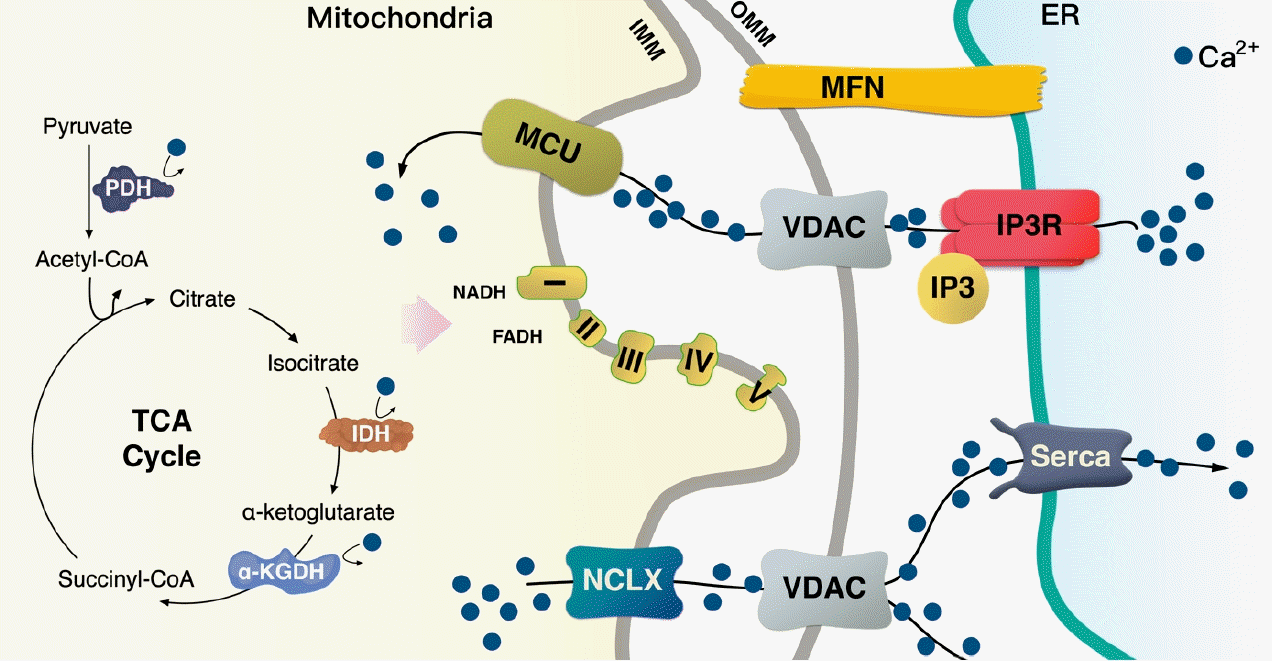
Ca2+ signaling and flux regulate numerous cellular physiological processes, including neuronal excitability, muscle contraction, nuclear gene expression, and mitochondrial integrity, function, and dynamics [27]. Accumulated data have shown that mitochondrial Ca2+ content is low under basal conditions, while increases in cytosolic free Ca2+ in response to various agents (nutrients, hormones, neurotransmitters, etc.) elevates mitochondrial Ca2+ levels [28,29]. This enhances the activity of tricarboxylic acid (TCA) cycle dehydrogenase (pyruvate-, isocitrate-, and α-ketoglutarate dehydrogenase) required for oxidative phosphorylation [30]. Thus, activation of oxidative metabolism via enhanced Ca2+ flux can increase the supply of redox cofactors, such as a reduced form of nicotinamide adenine dinucleotide (NAD) and flavin adenine dinucleotide (FAD) to drive ETC and adenosine triphosphate (ATP) synthesis (Fig. 2).
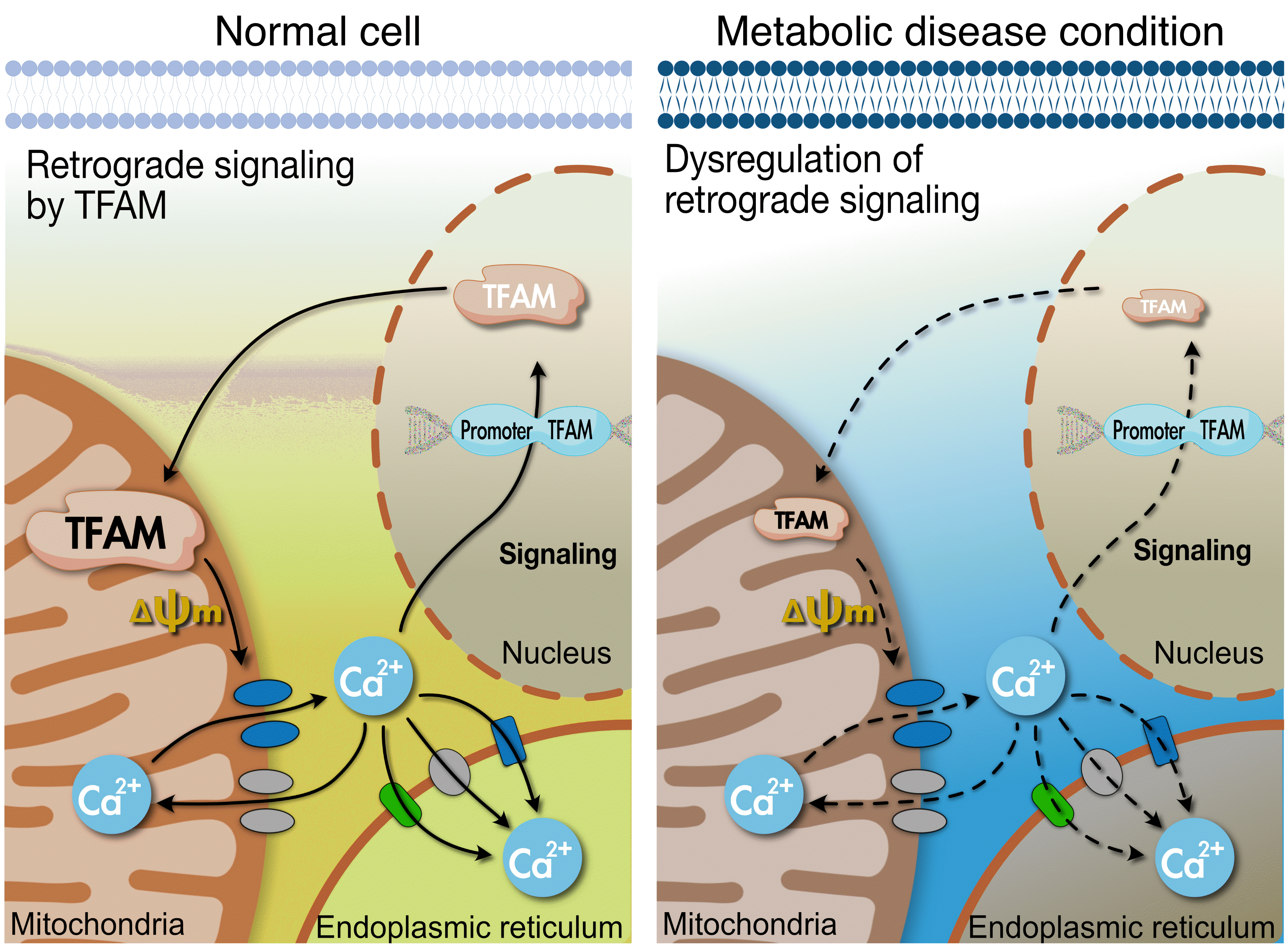
TFAM regulates the mitochondrial membrane potential (ΔΨm) and increases calcium/calmodulin-dependent protein kinase kinase β (CaMKKβ) [10]. In turn, ΔΨm can regulate cellular Ca2+ flux [31], while CaMKKβ is activated in a calcium-dependent manner [32]. Thus, TFAM-driven ΔΨm controls Ca2+ flux in the cell via various Ca2+ channels. Perturbation of TFAM expression cannot protect mtDNA [18], while damaged mtDNA has been reported to increase ER stress [33]. This suggests that TFAM may be involved in the mitochondria-ER interactions.
The lumen of the ER is the main storage site of Ca2+ and is constantly refilled via the sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) pump [34,35]. When skeletal muscle contraction is initiated, cytosolic Ca2+ concentration is rapidly increased by 3 to 4-fold, with the sarcoplasmic reticulum (SR), an ER in skeletal muscle, optimizing the coupling of excitation and contraction of muscle fibers. SR has channels for efflux of Ca2+ into the cytosol in response to depolarization, while SERCA transports cytosolic Ca2+ in an ATP-dependent manner into the ER lumen, thereby terminating the contraction. The close link between the mitochondria and SR/ER allows for rapid and potent local Ca2+ signaling (Fig. 2) [6,36].
Mitochondrial and endoplasmic reticulum contacts (MERC) orchestrate cellular physiological functions such as mitochondrial Ca2+ signaling and dynamics. Under physiological stimulation requiring intercommunication between mitochondria and ER, mitofusin 2 (MFN2) tethers to stabilize both organelles [31,37,38]. Meanwhile, inositol 1,4,5-trisphosphate (IP3) binds to the IP3 receptor (IP3R), triggering Ca2+ release from the ER through IP3R, which can directly release Ca2+ into the cytosol or mitochondria [36]. Released Ca2+ from the ER enters the mitochondrial intermembrane space by first passing through the outer mitochondrial membrane via the voltage-dependent anion channel (VDAC) [39], then the mitochondrial calcium uniporter (MCU) transporter across the inner mitochondrial membrane [40,41]. TFAM is linked to Ca2+ regulation via MFN2 [42]. Ca2+ flux is essential for mitochondrial bioenergetic processes, which is linked to the ΔΨm (Figs. 1 and 2).
TFAM regulates the mitochondrial and ER Ca2+ signaling to nucleus via ΔΨm
Mitochondria are key organelles in Ca2+ flux regulation in cells. While Ca2+ flux in mitochondria is one of the most significant processes in regulating energy production and communication with other organelles, it mediates various pathologies associated with metabolic disease.
Mitochondrial Ca2+ uptake is directly linked to mitochondrial bioenergetics, where depletion of mitochondrial ΔΨm abrogates mitochondrial Ca2+ uptake, and defects in the respiratory chain have been linked to the decreased ability of mitochondria to pump Ca2+. Energetic substrates derived by the TCA cycle are serially reduced to equivalents by ETC, and these redox reactions are coupled with protons pumping from the matrix into the intermembrane space [43]. The proton electrochemical gradient induced by ΔΨm and pH gradient is necessary to produce ATP (Fig. 1). Thus, ΔΨm can maintain mitochondrial Ca2+ uptake and physiological functions (Figs. 1 and 2) [44,45].
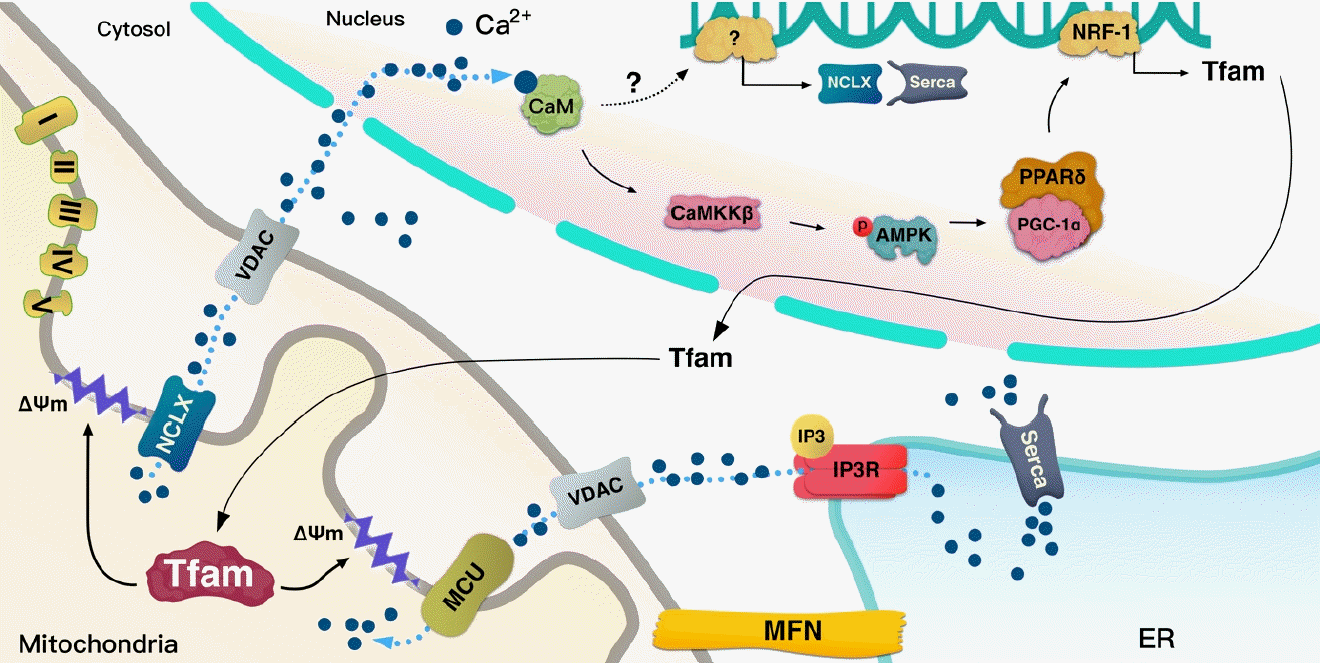
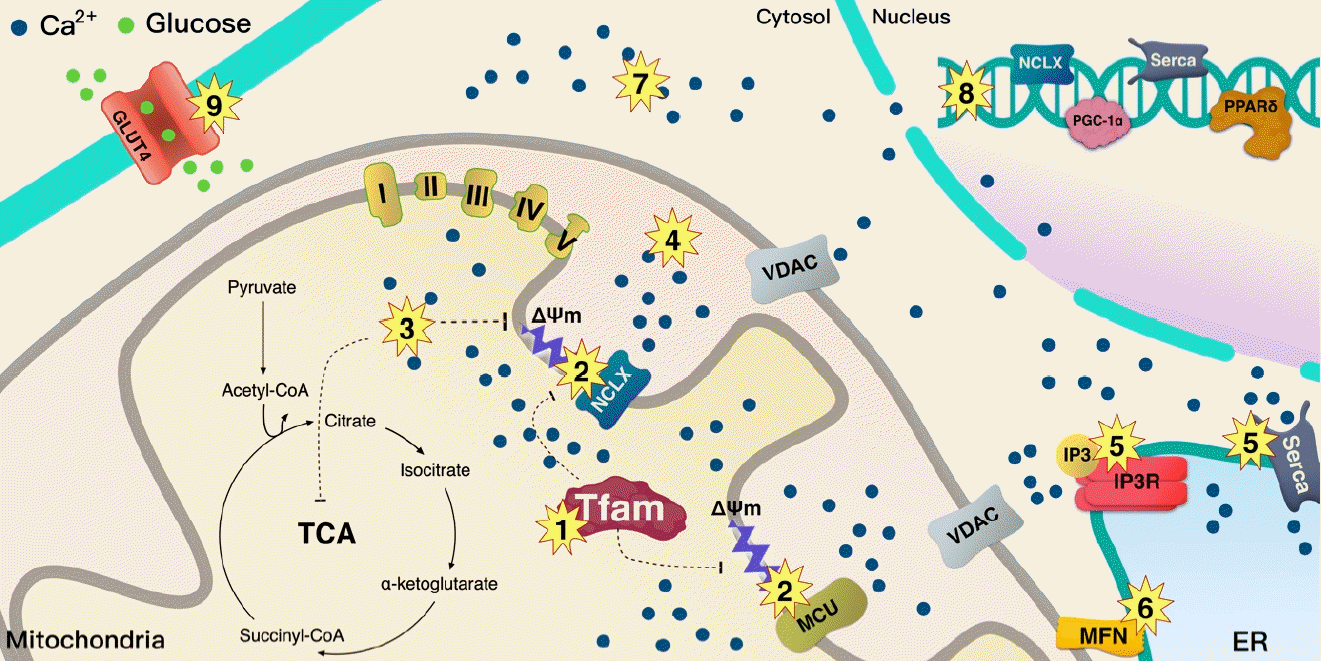
Lack of TFAM decreases Serca2a expression from the nucleus [46], indicating that TFAM in the mitochondria signals to the nucleus to transcribe Serca2a and coordinate with ER to maintain Ca2+ homeostasis. The ER can regulate the Ca2+ efflux channels [36], which could affect cytosolic Ca2+ and force during contraction; hence, a reduced ER Ca2+ level in TFAM knockout muscle results in a decrease in cytosolic Ca2+ [47], which leads to a lower force in single fibers during contraction [47]. In TFAM knockout mice, an increase in mitochondrial Ca2+ occurs during repeated contraction [47], which may induce Ca2+ overload in the mitochondria and oxidative stress. In contrast with TFAM knockouts, Serca2 gene can enhance contractile function and restore electrical stability in a heart failure model caused by ER(SR) Ca2+ leak [48]. Watanabe et al. [46] have shown that TFAM regulates Serca2 gene transcription; however, it is not clear how TFAM in mitochondria regulates nuclear Serca2a transcription (Fig. 3).
As mentioned above, mice with muscle-specific deletion of TFAM exhibit Ca2+ overload in the mitochondria, which might be caused by the low Ca2+ efflux into the cytosol by the mitochondria. MCU is the main channel transporting Ca2+ from the cytosol, and Na+/Ca2+ exchanger (NCLX) regulates efflux of Ca2+ from mitochondria, both of which are located at the inner mitochondrial membrane where they coordinate to regulate mitochondrial Ca2+ homeostasis. Mice with cardiac-specific deletion of TFAM showed how a failure in the coordination between MCU and NCLX in mitochondria can lead to cardiomyopathy [49]. TFAM deletion in cardiac muscle decrease Ca2+ efflux from mitochondria due to low NCLX gene and protein levels, with the reduced efflux potentially inducing mitochondrial Ca2+ overload [49]. Ca2+ flux through the MCU can be regulated by the voltage gradient across the inner mitochondrial membrane, while the opening probability of MCU is decreased based on ΔΨm depolarization [50]. Moreover, protonophore-induced depolarization of ΔΨm almost fully suppresses Ca2+ influx into the mitochondria [51]. These findings indicate that the large electrical driving force that arises from the negative potential across the inner ΔΨm is a major factor in regulating the influx of Ca2+ through MCU (Fig. 3).
Depolarization of ΔΨm or lack of mtDNA prevents mitochondrial Ca2+ uptake, elevating the cytoplasmic Ca2+ level and leading to mitochondrial retrograde signaling into the nucleus to activate Ca2+-mediated transcription mechanisms involved in calcineurin and Ca2+/calmodulin-dependent protein kinase (CaMK) [44,45]. Human TFAM (hTFAM) transgenic mice exhibit mild ΔΨm uncoupling when fatty acids are used as a substrate for skeletal muscle mitochondria, despite mtDNA in skeletal muscle being increased by hTfam [10]. These alterations by hTfam result in an increase in mild uncoupling in ΔΨm, leading to an increase in CaMKKβ expression from the nucleus [10]. Therefore, TFAM regulates ΔΨm and mediates retrograde signaling to the nucleus by Ca2+ transmission. Interestingly, mild uncoupling of ΔΨm by hTFAM decreased ROS [10]. Since a lack of TFAM can mediate Ca2+ overload in the cytoplasm, it not only induces retrograde signaling to the nucleus but also increases ROS and apoptosis [45]. ROS produced by lack of TFAM in cells may be induced by an overload of Ca2+ [45]; however, hTFAM overexpression can decrease ROS and oxidative stress in tissue with mild uncoupling of ΔΨm [10]. Therefore, TFAM may mediate mild uncoupling of ΔΨm that regulates Ca2+ retrograde signaling to tightly control cellular ROS (Fig. 3).
The nuclear genome encodes approximately 1,500 proteins that are necessary for mitochondrial function and integrity [52,53]. Intercommunication between mtDNA and the nuclear genome is necessary for mitochondrial biogenesis and normal function. To facilitate the interplay between the organelles, TFAM functions signaling transmitter to the mitochondria from the nucleus, and sends signals back to the nucleus according to the state of the mitochondria by regulating Ca2+. This signaling pathway is important for the regulation of normal mitochondrial biogenesis via peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), which orchestrates various transcription factors including NRF-1 and peroxisome proliferator-activated receptor delta (PPARδ) (Fig. 3).
Intracellular Ca2+ binds CaM, thereby mediating various cellular functions [3,54]. In skeletal muscle, this Ca2+/CaM complex contract the muscle fiber [55]. The activation of Ca2+/CaMKKβ enhances AMPK activation [32], which facilitates mitochondrial biogenesis via enhanced expression of PGC-1α [56]. Muscle-specific hTFAM transgenic mice exhibit higher levels of CaMKKβ and activated AMPK [10]. To maintain and manage mtDNA as well as to regulate mitochondrial function, it is also necessary to increase the amount of TFAM as the mitochondrial number increases. It has been shown that TFAM overexpression in skeletal muscle enhances the expression of NRF-1, which is an upstream transcription factor for itself [10] and PPARδ expression. PPARδ regulates glucose transporter type 4 (GLUT4) expression and glucose uptake in muscle tissue [57]. A higher level of PGC-1α and PPARδ in tissue is beneficial in improving metabolic diseases such as insulin resistance and T2DM (Figs. 3 and 4).
Ca2+ flux in metabolic disease
Skeletal muscle, liver, and adipose are metabolic and insulin-sensitive tissues. Mitochondrial dysfunction in those tissues is associated with lower levels of TFAM in various metabolic diseases such as obesity, insulin resistance, and T2DM, which mediate abnormal Ca2+ flux.
Role of MERC in diabetes
Tethering between mitochondria and ER plays a key role in Ca2+ homeostasis, which regulates energy metabolism, transportation of lipids, and apoptosis [58]. The influx of Ca2+ mediates insulin-stimulated glucose uptake in skeletal muscle [4]. The authors reported that a decrease in Ca2+ influx by IP3R inhibition improved insulin-stimulated glucose uptake in skeletal muscle without AKT signaling [4]. In adipocytes, cytosolic Ca2+ levels were increased by insulin stimulation [59]. The inhibition of Ca2+ signaling in adipocytes [59] and lack of IP3R in primary rat cardiomyocytes [60] decreased GLUT4 translocation and glucose uptake upon insulin stimulation, respectively. Moreover, mitochondrial dysfunction [61], dysregulation of lipid and Ca2+ homeostasis [62], and ER stress [63] have been reported to be closely linked to insulin resistance in the liver. Palmitate is known to reduce insulin sensitivity; it has been shown that disruption of the interaction between mitochondria and ER by palmitate induces insulin resistance in human’s and mice’s hepatocytes. However, an increase in these associations between organelles can prevent palmitate-induced insulin resistance [64]. Loss of IP3R1 decreases MERC formation and induces mitochondrial dysfunction and insulin resistance; however, restoration of MERC has been shown to improve palmitate-induced insulin resistance in hepatocytes [64]. It has been reported that AKT phosphorylates the IP3R channel, resulting in decreased Ca2+ release capacity through IP3R [65,66]. Moreover, loss of IP3R activity has been shown to change the integrity of MERC [64]. Although defects in MERC have been suggested to play a role in insulin resistance and T2DM [37,38,42], MERC-mediated Ca2+ flux for glucose homeostasis is complicated and tightly regulated; hence, other specific mechanisms might be involved in this process. Thus, further studies are required to demonstrate the relationship between insulin resistance and MERC, as well as the mechanisms involved in this process. ER-associated mitochondrial division sites are spatially linked to mitochondrial nucleoids, which suggests a specific role for mitochondria-ER contacts in mtDNA maintenance [67]. ER-mitochondria contacts coordinate mtDNA synthesis with division to distribute newly replicated nucleoids into daughter mitochondria [68]. MFNs, proteins acting between mitochondria and ER [69], are also involved in metabolic diseases. Mitochondrial dysfunction is reportedly caused by depletion and point mutations of mtDNA in mice with muscle-specific deletion of MFN1 and MFN2 [69], while MFN2 expression in skeletal muscle is lower in patients with obesity or T2DM [37]. In contrast, overexpression of MFN2 has been shown to improve diet-induced insulin resistance [70], and MFN2 is necessary for normal glucose homeostasis [38]. Whether TFAM is directly involved in the process of forming or maintaining ER-mitochondria contacts remains to be determined, however, the loss of MFN1 and MFN2 decreases mtDNA that is maintained by TFAM [69], while PGC-1α, a transcription factor for Tfam, can increase the MFN2 expression levels via estrogen-related receptor-α (ERRα) [71]. Moreover, TFAM is involved in packaging mtDNA into a nucleoid [72-74]; hence, it is possible that TFAM indirectly participates in this process of MERC-mediated glucose homeostasis. hTFAM overexpression in skeletal muscle has been reported to prevent high-fat diet-induced insulin resistance along with preserving higher levels of mtDNA [10].
SERCA tightly regulates cytosolic Ca2+, leading to glucose oxidation [75]. An increase in SERCA has been reported to improve diabetic cardiomyopathy [7,76]. A high-fat diet decreases Serca2a expression due to a lack of TFAM; however, overexpression of TFAM inhibits hydrogen peroxide-induced decrease of Serca2a mRNA [46]. An increase in SERCA2a by TFAM can reduce levels of cytoplasmic Ca2+ and calpain and mitochondrial apoptosis factor, thereby improving glucose uptake in cardiac muscle [77]. Increased expression of Serca improves metabolic syndrome [78].
Role of mitochondrial Ca2+ regulation in T2DM
The MCU complex consists of a pore-forming MCU subunit, a regulatory subunit, and mitochondrial Ca2+ uptake proteins 1-3 (MICU1-3), which regulate the channel activity [79-82]. Indeed, a splicing variant of muscle-specific MICU1 with higher Ca2+ affinity facilitates mitochondrial Ca2+ uptake and ATP production, which is required for muscle contraction [83], whereas a deletion of MCU in skeletal muscle in mice decreases muscle force, indicating that through regulating Ca2+ uptake, MCU plays a vital role in energy production for contraction [84]. Deletion of the muscle-specific MCU induced lower activity of pyruvate dehydrogenase (PDH), which is sufficient to shift the preference of the substrate toward fatty acids from carbohydrates [85] since PDH converts pyruvate to acetyl-CoA. In addition, loss of MCU causes defects in Ca2+-sensitive TCA cycle enzymes such as isocitrate and α-ketoglutarate dehydrogenases. Therefore, MCU-deleted mitochondria in skeletal muscle cannot sustain respiration without TCA cycle support, although MCU-deleted muscle mostly relies on fatty acids as substrate for mitochondrial respiration [85]. Thus, fatty acids do not completely oxidize and accumulate in muscle, leading to insulin resistance in skeletal muscle [86]. MCU is on the mitochondrial inner membrane that is dependent on ΔΨm, and hTFAM overexpression in skeletal muscle induces mild uncoupling of ΔΨm, which may change mitochondrial Ca2+ flux. This alteration shifts the preferred substrate for mitochondrial respiration from glucose to fatty acids [10]. However, the hTFAM transgenic model is different from the MCU deletion since muscle-specific hTFAM overexpression increases glucose uptake and prevents high-fat diet-induced insulin resistance (Figs. 3 and 4) [10].
The NCLX plays a vital role in mitochondrial efflux into the cytosol [87]. A decrease in NCLX function leads to the accumulation of Ca2+ in mitochondria, while impaired NCLX function has been reported in diabetic rat hearts [87,88]. A previous study has shown that deletion of TFAM in mouse cardiomyocytes decreases NCLX transcription [49], and TFAM regulates ΔΨm in skeletal muscle; therefore, TFAM seems to clearly link to mitochondrial Ca2+ efflux via ΔΨm (Figs. 3 and 4).
Overloaded Ca2+ levels have been reported in adipocytes from patients with obesity with insulin resistance [89] and from T2DM rats [90], which may link to the abnormal functions of mitochondria and ER which are unable to maintain Ca2+ homeostasis in cells. ΔΨm depolarization enhances ROS production [91-93]; however, hTFAM overexpression prevents fatty acid-induced ΔΨm depolarization in muscle cells and blocks ROS production in mouse skeletal muscle [10]. Moreover, mild ΔΨm uncoupling induced by hTFAM increases glucose uptake in skeletal muscle and improves high-fat diet-induced insulin resistance [10]. Further studies are required to determine how hTFAM regulates Ca2+ flux in mitochondria via ΔΨm.
CONCLUSIONS
This review has provided an overview of how mitochondria signaling to the nucleus and interact with the ER to regulate Ca2+ as well as the role of mitochondrial dysregulation via cellular Ca2+ homeostasis in the pathogenesis of metabolic diseases, including insulin resistance and T2DM. Although further investigation is required to determine mechanisms by which TFAM controls ΔΨm to regulate Ca2+ flux in the cell, accumulated data indicate that TFAM-driven ΔΨm regulates mitochondrial and ER’s Ca2+ flux. This leads to Ca2+ signaling into the nucleus, thereby inducing the expression of various genes such as flux channels and signaling co-factors for Ca2+. TFAM-mediated regulation of ΔΨm prevents high-fat diet-induced oxidative stress and insulin resistance via enhanced expression of GLUT4, PGC-1α, and PPARδ from the nucleus. Moreover, the mitochondria interact with the ER and regulate cellular Ca2+ flux. This process clearly influences mitochondrial TCA cycle and oxidative phosphorylation, whereas dysregulation of this process could increase metabolic diseases such as T2DM. Several fundamental cellular processes are governed by this crosstalk among the mitochondria, ER, and nucleus, which plays an essential role in the regulation of the cellular metabolic response to environmental cues. The blockage of this communication reduces mitochondrial activity [94,95] and increases the incidence rate of metabolic disease. Thus, the mitochondria govern intercommunication between cellular organelles, including ER and nucleus, via Ca2+ signaling to regulate metabolism.
Taken together, here, we propose that TFAM is involved in the regulation of Ca2+ flux via the mitochondria-ER interaction that can signal to the nucleus, which ultimately mitigates metabolic disorders.
 XML Download
XML Download