

 PDF
PDF Citation
Citation Print
Print



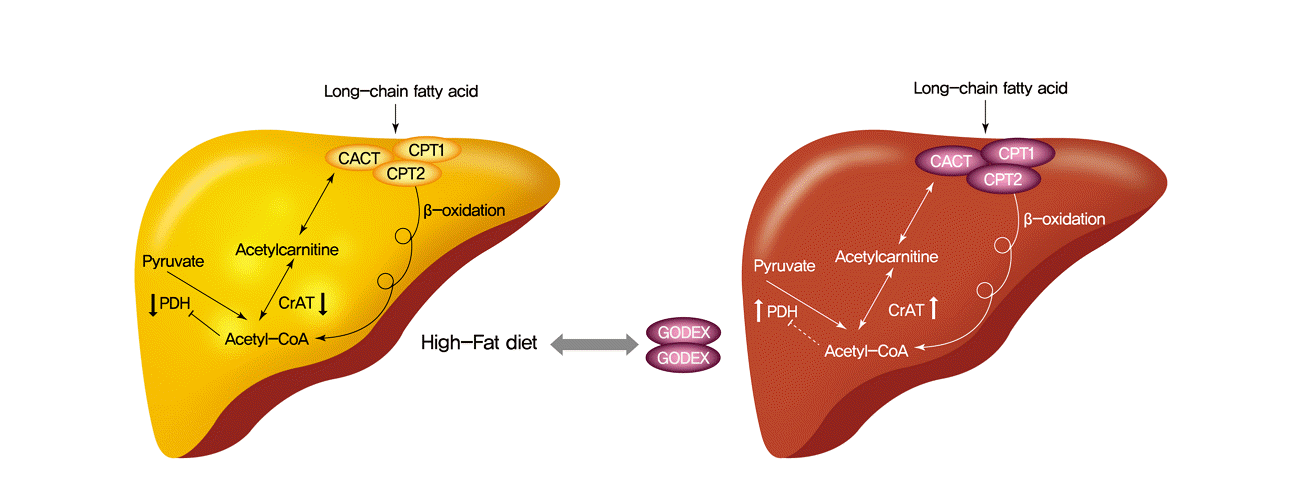
INTRODUCTION
Non-alcoholic fatty liver disease (NAFLD) is closely related to obesity, hyperlipidemia, and type 2 diabetes mellitus (T2DM). The primary pathogenesis is insulin resistance, which could result in several changes in lipid metabolism, such as enhanced lipolysis, increased triglyceride (TG) synthesis, and hepatic uptake of fatty acids [1]. Each of these might contribute to mitochondrial dysfunction and accumulation of hepato-cellular TG [2], which could in turn result in a preferential shift from carbohydrate to fatty acid oxidation [2,3]. The liver is a central organ that regulates glucose metabolism by controlling glycogen synthesis and gluconeogenesis; however, if not adequately controlled, it could further aggravate glucose and lipid metabolism [4].
The carnitine orotate complex (Godex) is a hepatoprotector prescribed to normalize alanine aminotransferase (ALT) levels [5] and biochemical parameters [6] and inhibits hepatocyte necrosis, and could also improve severe hepatocellular vacuolar degeneration, lobular restructuring and necrosis of bile duct in CCl4- and ethanol-induced liver injuries [7], and decreased urinary 8-hydroxy-2’-deoxyguanosine levels and increased mitochondrial DNA (mtDNA) copy number in patients with impaired glucose metabolism and fatty liver [8]. We have previously reported that Godex had beneficial effects on serum ALT, fasting glucose, glycosylated hemoglobin level, homeostatic model assessment for insulin resistance (HOMA-IR), and significantly improved hepatic steatosis in patients with T2DM and NAFLD [9]. Muoio et al. [10] have shown that muscle-specific loss of carnitine acetyltransferase (CrAT) in mice led to an accumulation of acetyl-coenzyme A (CoA) within the mitochondria and allosteric inhibition of pyruvate dehydrogenase (PDH), which subsequently impaired glucose utilization. Seiler et al. [11] proposed that CrAT activity decreased in response to genetic diabetes, high-fat diet (HFD), and lipid exposure. The favorable effects of carnitine, one of the components of Godex, on muscle glucose metabolism through the modulation of the intramitochondrial acetyl-CoA/CoA ratio and the activity of the PDH complex and stimulation of the insulin-like growth factor-1 (IGF-1) signaling cascade were also reported [10,12]. Considering the vital role of each of these mechanisms in muscle, we hypothesized that Godex would improve exaggerated metabolic dysregulation through the control of acetyl-CoA in liver. This study was conducted to investigate the effects of Godex in the regulation of glucose and lipid metabolism through CrAT-mediated pathways in a HFD-fed mice.
Go to : 
METHODS
Animal study
Male C57BL/6J mice aged 4 weeks were obtained from The Jackson Laboratory (JAX, Bar Harbor, ME, USA) and housed in 25°C temperature and 12-hour light-dark cycle. The animals were fed a normal-fat diet (ND, 10% fat calories) or HFD (60% fat calories) (Research Diets Inc., New Brunswick, NJ, USA) ad libitum for 8 weeks. Food intake was measured every 24 hours to derive food consumption. Caloric content of food intake was determined based on 3.573 kcal/g for ND and 5.404 kcal/g for HFD. Godex was provided by CelltrionPharm (CelltrionPharm Inc., Seoul, Korea). HFD mice were subdivided into vehicle-treated phosphate-buffered saline, Godex 500 mg/kg/body weight (B.W.) (G500) and Godex 2,000 mg/kg/B.W. (G2000) groups. Body weight and food intake were measured weekly. This study was reviewed and approved (No. 2017-0228001) by the Institutional Animal Care and Use Committee of Samsung Biomedical Research Institute, which is an Association for the Assessment and Accreditation of Laboratory Animal Care International (AALAC)-accredited facility and abides by the Institute of Laboratory Animal Resources guide. After 7 weeks, mice were administered either D-glucose (2 g/kg/B.W.) or human insulin (0.75 units/kg/B.W.) for oral glucose tolerance test or insulin tolerance test, respectively. Blood samples were obtained from tail tips to measure glucose levels at the appropriate time points using a glucometer (AccuCheck II; Roche, Indianapolis, IN, USA). Kitt is the first-order rate constant for the disappearance of glucose (Kitt) during the 3 to 15 minutes period after intravenous insulin administration, and was taken as a measure of insulin sensitivity, and was calculated as follows: Kitt (%/min)=(0.693×100)/t1/2, where t1/2 is the “half-life” calculated from the slope of the plasma glucose concentration. At the end of experiments, abdominal adipose tissue quantification was performed using the Siemens Inveon Micro-CT scanner (Siemens Healthcare, Malvern, PA, USA). Low-density lipoprotein cholesterol (LDL-C) level was calculated by the Friedewald equation [13]. Serum levels of adiponectin (Millipore, Danvers, MA, USA), ALT, aspartate aminotransferase (AST) (Asan Pharm., Seoul, Korea), and insulin (Mercodia, Uppsala, Sweden) were determined, and the insulin sensitivity index (HOMA-IR=glucose×insulin/22.5) was also calculated.
Indirect calorimetry
Oxygen consumption (VO2) and carbon dioxide output (VCO2) were measured (Oxylet; Panlab-Bioseb, Vitrolles, France) during a 24-hour period. Respiratory quotient (RQ), energy expenditure, glucose oxidation, and lipid oxidation were calculated [14].
Histological analysis, electron microscopy, and lipid contents of the liver
The hepatic tissues were stained with H&E and examined using light microscopy. Liver steatosis stages were evaluated semiquantitatively according to the NASH-Clinical Research Network criteria [15]. The amount of steatosis (percentage of hepatocytes containing fat droplets) was scored as 0 (<5%), 1 (5%–33%), 2 (>33%–66%), and 3 (>66%). Liver sections were fixed with 2.5% glutaraldehyde and 4% paraformaldehyde, then stained with lead-uranyl acetate, and changes in mitochondrial morphology were observed with a Hitachi HT7700 electron microscope (Hitachi, Tokyo, Japan). Lipid extraction was performed using the Folch et al.’s method [16]. Hepatic TG and cholesterol contents were measured using commercially available kits (Wako Pure Chemical Industries, Osaka, Japan). Malonyl-CoA concentration was determined using the kit (Cusabio, Wuhan, China).
OXPHOS enzyme activity, CrAT assay, acetyl-CoA and CoA measurements, and PDH activity
Oxidative phosphorylation (OXPHOS) complexes was measured with the MitoToxTM Complete OXPHOS Activity Assay kit (Abcam, Cambridge, MA, USA). CrAT was measured with a CrAT assay kit (USCN Life Science, Wuhan, China). Concentration of acetyl-CoA and CoA and PDH activity were determined using the respective kits (BioVision, Milpitas, CA, USA).
Gene silencing
Hepa-1c1c7 cells were transfected with small interfering RNA (siRNA) of carnitine palmitoyltransferase-1 (CPT-1), CPT-2, carnitine acylcarnitine translocase (CACT) and CrAT (Santa Cruz Biotechology, Santa Cruz, CA, USA) using LipofectamineRNAiMAX (Invitrogen, Waltham, MA, USA), and replaced with or without 800 μM palmitic acid (PA) with Godex after 24 hours of transfection. Four-week-old male C57BL/6J mice were fed ND or HFD with or without carnitine, the main component of the Godex (10 g/kg B.W., intraperitoneal injection), for 8 weeks. A 50 μg siCrAT in 1 mL saline was injected into the tail vein for 2 days and then sacrificed. Changes in genes of β-oxidation and gluconeogenesis were observed in liver tissue.
Cell culture
Human hepatocellular carcinoma cells (HepG2) were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) medium and mouse hepatoma cells (Hepa-1c1c7) were maintained in alpha modification of Eagle’s minimum essential media (α-MEM) containing 10% fetal bovine serum and 1% antibiotics in 5% CO2 humidified atmosphere at 37°C, respectively. Cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA), and all chemicals used in cell culture were obtained from Sigma-Aldrich (St. Louis, MO, USA) unless otherwise indicated.
mRNA, DNA, and protein analyses
Quantitative reverse-transcription polymerase chain reaction (PCR) was carried out as described previously [17]. Genomic DNA was used to analyze the mtDNA/nuclear DNA (nDNA) ratio by absolute quantification using PCR for mouse mtDNA (12S, Cox1) and mouse nDNA (hypoxanthine phosphoribosyltransferase [HPRT]). The Western blots were visualized with ECL Plus Western Blotting Detection System (GE Healthcare, Madison, WI, USA).
Visualization of mitochondria
Mitochondria were identified by MitoTracker Red (Molecular Probes Inc., Eugene, OR, USA) staining [18] and analyzed with a Leica confocal laser scanning microscope using SCANware software (Leica Lasertechnik GmbH, Heidelberg, Germany).
Statistical analyses
Results were expressed as mean±standard deviation and were analyzed using SPSS version 10.0 software (SPSS Inc., Chicago, IL, USA). Data were validated by analysis of variance (ANOVA), and a P value <0.05 as indicated by Duncan’s multiple range test was considered to indicate a statistically significant difference.
Go to :
RESULTS
Godex prevented diet-induced obesity
After 8 weeks, there was a 64% increase in body weight in the HFD group compared to the ND group. Godex dose-dependently decreased body weight than the HFD group by 22% and 36% at 8 weeks, respectively (Fig. 1A). The HFD group showed significant increases in visceral and subcutaneous fat volume relative to the ND group, which were normalized in the G2000 group (Fig. 1B). These alterations were not due to differences in food intake, as food consumptions were similar among all groups (Fig. 1C). Compared to the ND group, the energy intake of the HFD-supplied groups increased, but there was no significant difference between the groups (Fig. 1D). G2000 group showed significant increase in plasma adiponectin levels compared with the HFD group (Fig. 1E). RQ was significantly higher in the G2000 group than the HFD group (Fig. 1F). These results were confirmed by the amount of glucose oxidized (Fig. 1G), which was higher in the G2000 treated group than the HFD group, whereas the amount of lipids oxidized (Fig. 1G) and energy expenditure (Fig. 1H) did not differ among the HFD-treated oups.
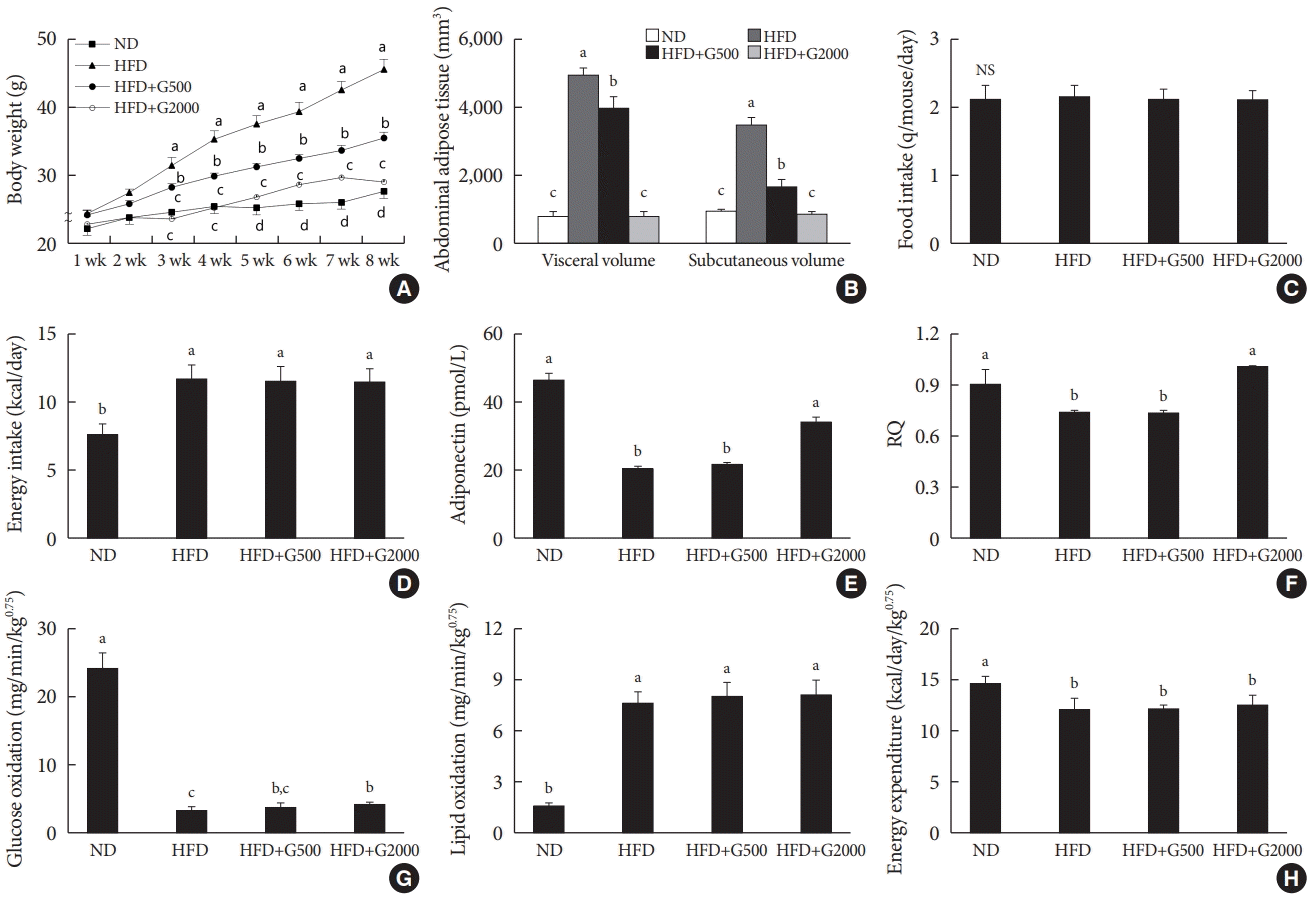 | Fig. 1.Carnitine orotate complex (Godex) prevented diet-induced obesity (A) body weight, (B) visceral and subcutaneous fat volume, (C) food intake, (D) energy intake, (E) plasma adiponectin levels, (F) respiratory quotient (RQ), (G) glucose oxidation and lipid oxidation, (H) energy expenditure in C57BL/6J mice during 8 weeks of normal-fat diet (ND) or high-fat diet (HFD) feeding. HFD mice were subdivided into vehicle-treated (HFD), Godex 500 mg/kg/body weight (B.W.) (HFD+G500), and Godex 2000 mg/kg/B.W. (HFD+G2000) groups. RQ was calculated as the ratio of carbon dioxide output (VCO2) to oxygen consumption (VO2). Energy expenditure (in kcal/day/kg0.75=1.44×VO2×[3.815+1.232×respiratory exchange ratio, RER]), glucose oxidation (in mg/min/kg0.75=[(4.545×VCO2)–(3.205×VO2)]/1,000), and lipid oxidation (in mg/min/kg0.75=[1.672×(VO2–VCO2)/1,000]) were calculated [14]. Values are presented as mean±standard deviation (n=10). NS, not significant. a,b,cValues within groups indicate significant difference at P<0.05.
|
Godex alleviated hepatic steatosis
As compared with ND group, the HFD group exhibited severe steatosis demonstrated by hepatic vacuoles, lipid droplets and hepatocyte swelling. Godex-supplemented groups showed remarkable reductions in the number and size of lipid droplets in the hepatic tissue (Fig. 2A) which were supported by the analysis of steatosis histology scores (Fig. 2B). Quantitative assay of lipid content showed significantly increased hepatic TG and cholesterol (Fig. 2C) in the HFD group, which were significantly reduced by Godex. Liver injury of HFD group was confirmed by statistically significant increases of serum ALT (Fig. 2D) and AST (Fig. 2E), which were significantly reduced by Godex.
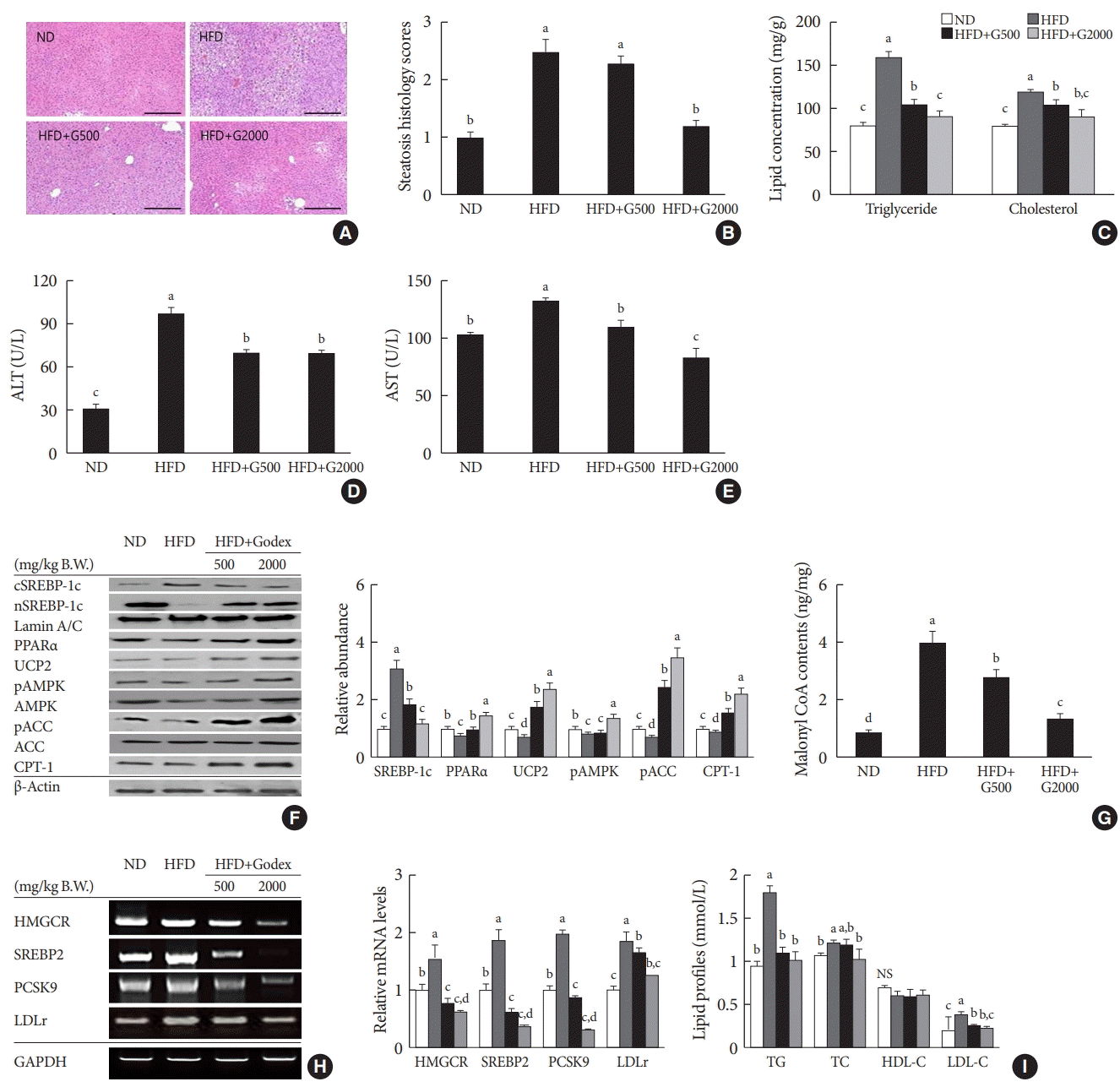 | Fig. 2.Carnitine orotate complex (Godex) alleviated hepatic steatosis. (A) H&E staining of the liver (×400), (B) steatosis histology scores, (C) hepatic lipid concentration, (D) alanine transaminase (ALT) levels, (E) aspartate aminotransferase (AST) levels, (F) β-oxidation-related protein expression in the liver, (G) hepatic malonyl coenzyme A (CoA) content, (H) hydroxymethylglutarylCoA reductase (HMGCR), sterol regulatory element-binding protein-2 (SREBP2), proprotein convertase subtilisin/kexin type 9 (PCSK9), and low-density lipoprotein receptor (LDLr) mRNA expression in the liver, (I) serum lipid profiles in C57BL/6J mice during 8 weeks of normal-fat diet (ND) or high-fat diet (HFD) feeding. HFD mice were subdivided into vehicle-treated (HFD), Godex 500 mg/kg/body weight (B.W.) (HFD+G500), and Godex 2,000 mg/kg/B.W. (HFD+G2000) groups. Data are presented as mean±standard deviation (n=10). PPARα, peroxisome proliferator-activated receptor alpha; UCP2, uncoupling protein 2; pAMPK, phosphorylated 5’AMP-activated protein kinase; pACC, phosphorylated acetyl-CoA carboxylase; CPT-1, carnitine palmitoyltransferase-1; GAPDH, glyceraldehyde 3 phosphate dehydrogenase; NS, not significant; TG, triglyceride; TC, total cholesterol; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol. a,b,c,dValues within groups indicate significant difference at P<0.05.
|
The translocation of sterol regulatory element-binding protein-1 (SREBP-1) from cytoplasm to the nucleus was inhibited by Godex (Fig. 2F). Expressions of lipolytic proteins, such as peroxisome proliferator-activated receptor alpha (PPARα), uncoupling protein 2 (UCP2), and CPT-1 were decreased in the HFD group than ND group but was significantly increased by Godex (Fig. 2F). The HFD group showed a significantly higher malonyl-CoA content than the ND group, but Godex dose-dependently decreased malonyl-CoA content (Fig. 2G). Godex also dose-dependently increased 5’AMP-activated protein kinase (AMPK) and acetyl-CoA carboxylase (ACC) phosphorylation compared to the HFD group (Fig. 2F). Hepatic mRNA levels of the SREBP target genes, namely, hydroxymethylglutaryl-CoA reductase (HMGCR), SREBP2, proprotein convertase subtilisin/kexin type 9 (PCSK9) and low-density lipoprotein receptor (LDLr) were significantly increased in the HFD group relative to the ND group but were significantly decreased by Godex (Fig. 2H). Serum TG, total cholesterol, and LDL-C levels were significantly increased in the HFD group than the ND group, which was normalized by Godex (Fig. 2I).
Godex improved mitochondrial function and biogenesis
Enzyme activities of the OXPHOS complexes were significantly reduced in the HFD group relative to the ND group but were significantly increased by Godex supplementation (Fig. 3A). HFD mice showed mitochondrial swelling, and cristae disorganization; however, Godex restored regular mitochondrial size and the cristae, with the typical folded intermembrane space and a dense matrix (Fig. 3B). Protein levels of sirtuin (SIRT1), peroxisome proliferator-activated receptor-gamma coactivator 1-alpha (PGC1α), and nuclear respiratory factor 1 (NRF1) were upregulated by Godex compared with the HFD group (Fig. 3C). To investigate whether Godex-fed mice showed alterations in mitochondrial biogenesis, we analyzed the mtDNA content (mitochondrial to nuclear gene [mt/nDNA] ratio), showed that Godex increased the mt/nDNA ratio in a dose-dependent manner (Fig. 3D). Moreover, MitoTracker Red staining intensity in HepG2 cells was significantly lower in PA-treated cells than control cells; however, Godex treatment increased the intensity by 33% compared to PA treatment (Fig. 3E). Protein levels of PGC1α, NRF1, and SIRT1 were upregulated by Godex in HepG2 cells (Fig. 3F).
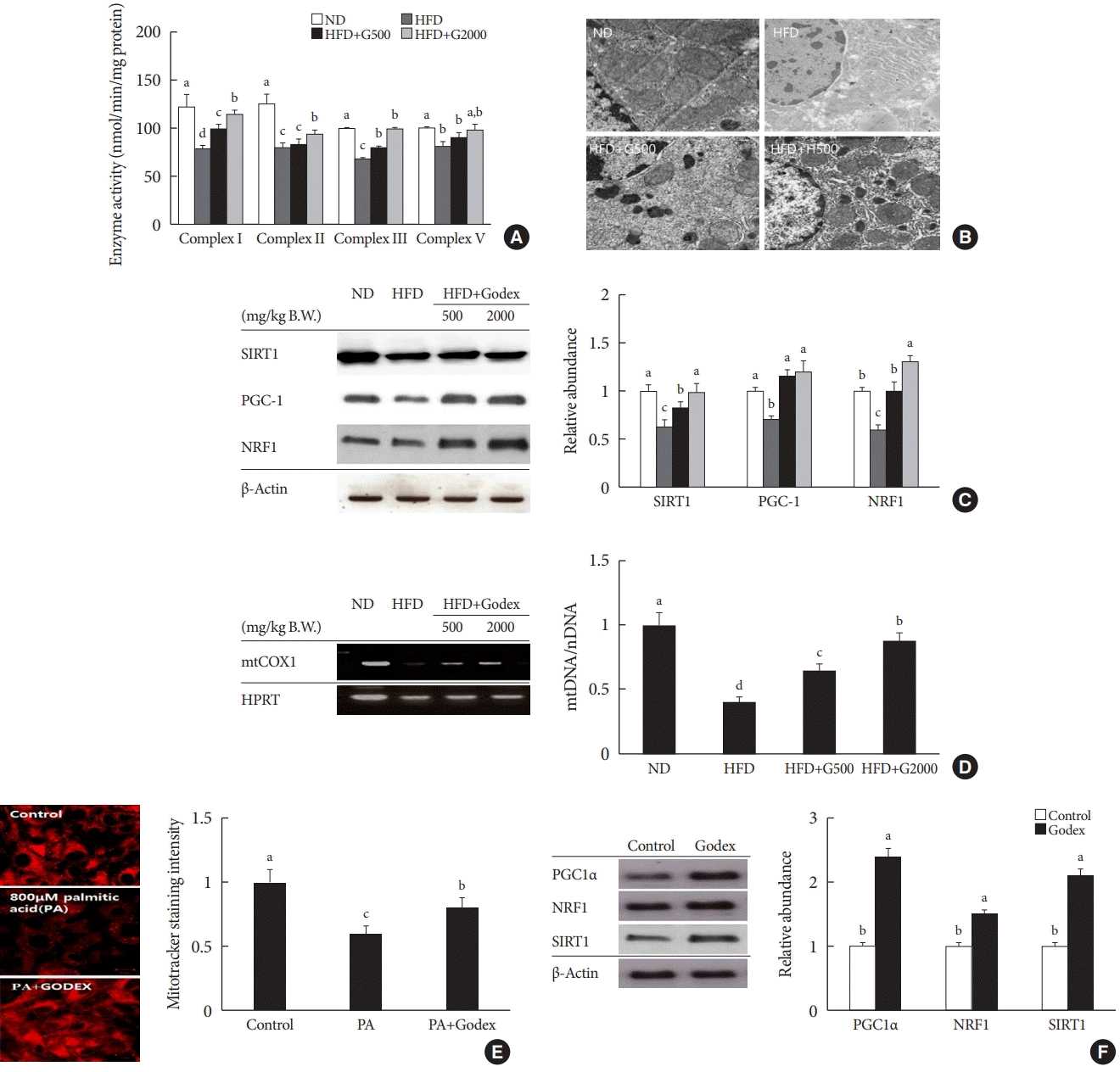 | Fig. 3.Carnitine orotate complex (Godex) improved mitochondrial function. (A) Hepatic oxidative phosphorylation (OXPHOS) enzyme activity, (B) transmission electron microscope images of liver mitochondria (×15,000) (C) proteins expression in mitochondrial metabolism in the liver (D) mitochondrial contents in C57BL/6J mice during 8 weeks of normal-fat diet (ND) or high-fat diet (HFD) feeding. HFD mice were subdivided into vehicle-treated (HFD), Godex 500 mg/kg/body weight (B.W.) (HFD+G500), and Godex 2,000 mg/kg/B.W. (HFD+G2000) groups. Scale bar: 40 μm. To correct for mitochondrial volume, all OXPHOS enzyme activities were normalized by the activity of citrate synthase. Data are presented as mean±standard deviation (n=10). (E) mitoTracker® staining intensity in HepG2 cells after treatment with 800 μM palmitic acid (PA) with 150 μg/mL Godex for 24 hours (×400). (F) proteins expression in mitochondrial metabolism in HepG2 cells after treatment with 150 μg/mL Godex for 24 hours. β-Actin was used as the loading control for normalization. Data are presented as mean±standard deviation (n=10). SIRT1, sirtuin 1; PGC1, peroxisome proliferator-activated receptor-gamma coactivator 1; NRF1, nuclear respiratory factor 1; mtCOX1, mitochondrial cytochrome c oxidase subunit 1; HPRT, hypoxanthine phosphoribosyltransferase. a,b,c,dValues within groups indicate significant difference at P<0.05.
|
Godex improved glucose tolerance and insulin sensitivity
Fasting blood glucose level (Fig. 4A), serum insulin (Fig. 4B), and C-peptide (Fig. 4C) levels were significantly higher in the HFD group than the ND group but were normal in the G2000 group. The average area under the curve (AUC) value was significantly higher in the HFD group than the ND group, and Godex supplementation significantly decreased the AUC compared to the HFD group, indicating an improvement in glucose tolerance (Fig. 4D). Kitt revealed a reduction in insulin sensitivity in the HFD group compared to the ND group but was normalized in G2000 group (Fig. 4E). The HOMA-IR of the HFD group was 4.3-fold higher than the ND group, and normal in the G2000 group (Fig. 4F). The expressions of phosphoenol pyruvate carboxykinase (PEPCK) and glucose 6-phosphatase (G6Pase) were increased in the HFD group compared to the ND group but were significantly decreased by Godex (Fig. 5A). CrAT content of the liver was reduced by 16% in the HFD group compared to the ND group (P<0.05) and was improved by G2000 supplementation (Fig. 5B). CrAT deficiency in HFD group resulted in an elevated mitochondrial level of acetyl-CoA (Fig. 5C) and a complete loss of PDH activity than the ND group (Fig. 5D). Hepatic β-hydroxybutyrate was significantly higher in the HFD group than the ND group but was significantly decreased by G2000 supplementation (Fig. 5E). Phosphorylations of insulin receptor β (IRβ), insulin receptor substrate 1 (IRS1) tyrosine, and Akt serine were increased in a dose-dependent manner by Godex (Fig. 5F). We also confirmed the positive effect of carnitine, the main component of Godex on β-oxidation and gluconeogenesis in the liver and HOMA-IR of HFD-fed mice, which were reversed by treatment with siCrAT (Fig. 5G-L).
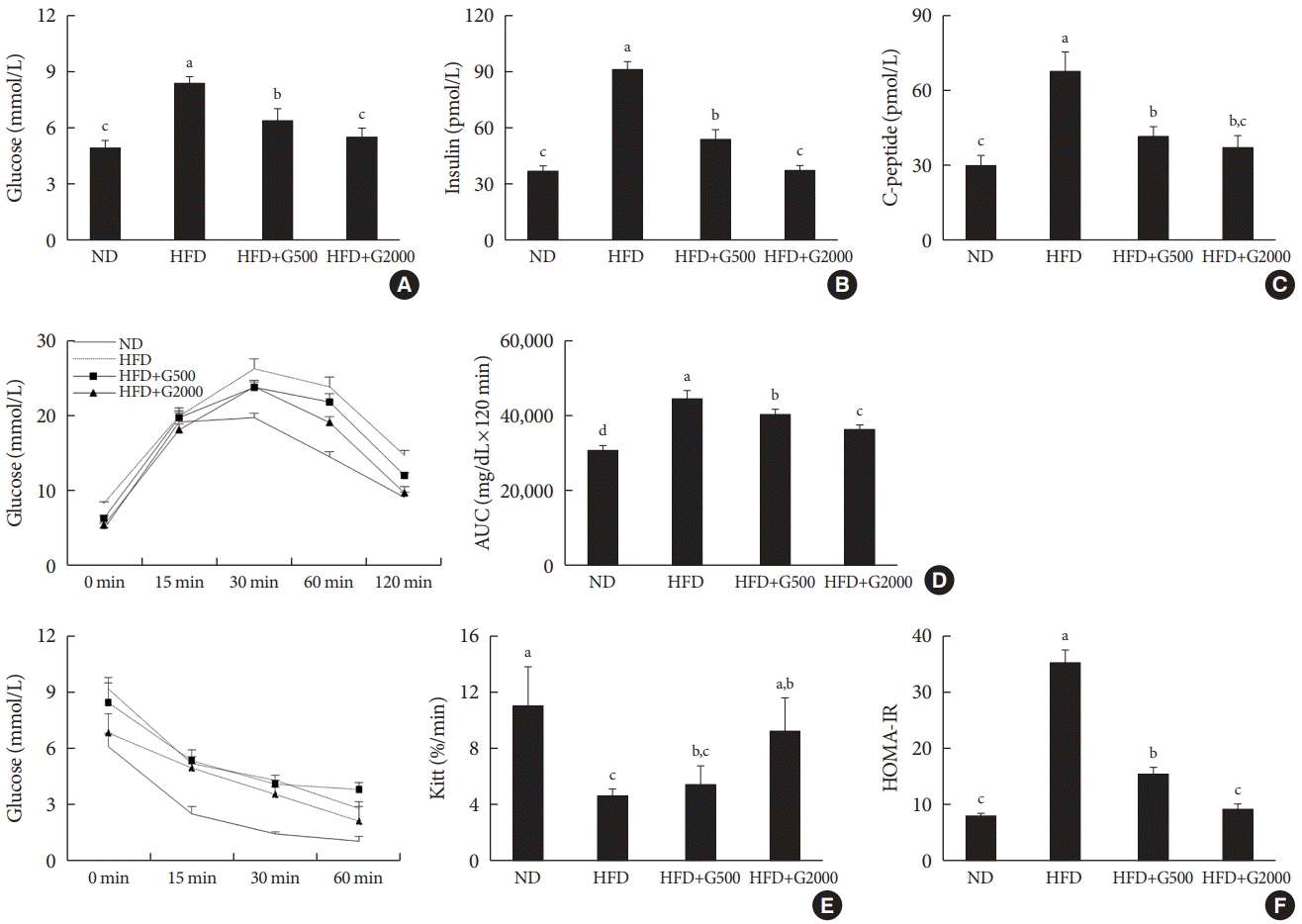 | Fig. 4.Carnitine orotate complex (Godex) improved glucose tolerance and insulin sensitivity. (A) Glucose levels, (B) insulin levels, (C) C-peptide levels, (D) oral glucose tolerance test and area under the curve (AUC), (E) insulin tolerance test (ITT) and Kitt, (F) homeostatic model assessment for insulin resistance (HOMA-IR) in C57BL/6J mice during 8 weeks of normal-fat diet (ND) or high-fat diet (HFD) feeding. HFD mice were subdivided into vehicle-treated (HFD), Godex 500 mg/kg/body weight (B.W.) (HFD+G500), and Godex 2,000 mg/kg/B.W. (HFD+G2000) groups. Data are presented as mean±standard deviation (n=10). a,b,c,dValues within groups indicate significant difference at P<0.05.
|
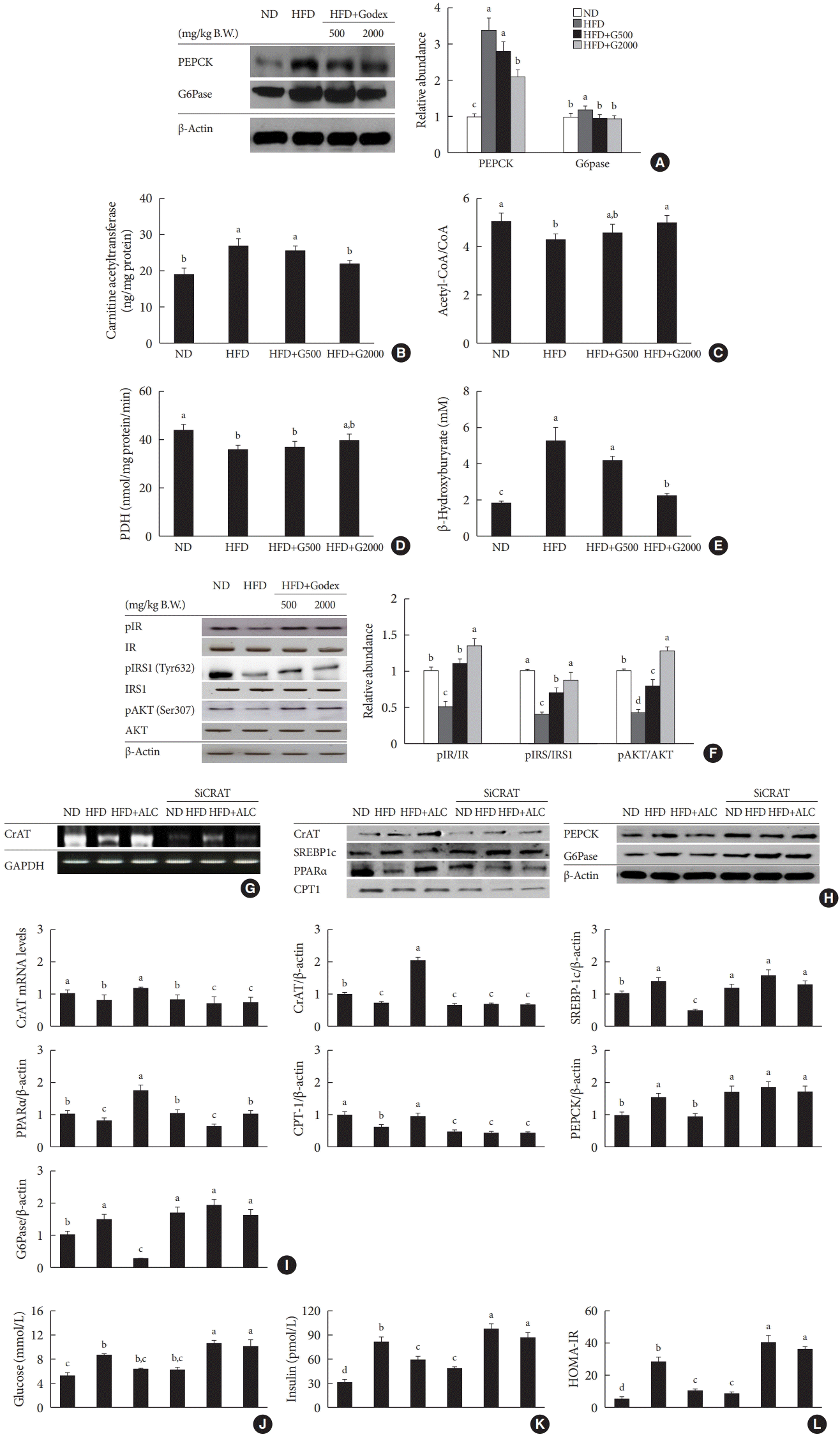 | Fig. 5.Carnitine ameliorated gluconeogenesis, a carnitine acetyltransferase (CrAT)-mediated pathway and insulin signaling in liver. (A) Protein levels of gluconeogenic genes, (B) hepatic mitochondrial CrAT content, (C) hepatic mitochondrial acetyl-coenzyme A(CoA)/CoA ratio, (D) hepatic mitochondrial pyruvate dehydrogenase (PDH) activity, (E) hepatic β-hydroxybutyrate, (F) hepatic insulin signaling in C57BL/6J mice during 8 weeks of normal-fat diet (ND) or high-fat diet (HFD) feeding. HFD mice were subdivided into vehicle-treated (HFD), carnitine orotate complex (Godex) 500 mg/kg/body weight (B.W.) (HFD+G500), and Godex 2,000 mg/kg/B.W. (HFD+G2000) groups. The effectiveness of CrAT siRNA knockdown was determined by (G) reverse transcription polymerase chain reaction (RT-PCR), (H) Western blotting. (I) CrAT mRNA level, and levels of CrAT, sterol regulatory element-binding protein-1 (SREBP-1), peroxisome proliferator-activated receptor alpha (PPARα), carnitine palmitoyltransferase-1 (CPT-1), phosphoenolpyruvate carboxykinase (PEPCK), and glucose 6-phosphatase (G6Pase) were assessed by Western blotting or (J) glucose levels, (K) insulin levels, and (L) homeostatic model assessment for insulin resistance (HOMA-IR). Four-week-old male C57BL/6J mice were fed a ND or HFD with or without carnitine, the main component of the Godex (10 g/kg B.W., intraperitoneal injection) for 8 weeks. Carnitine was used as acetyl-L-carnitine (ALC). Injected into the tail vein with 50 μg siCrAT in 1 mL saline over 2 days and then sacrified. Data are presented as mean±standard deviation (n=10). pIR, phosphorylated insulin receptor; pIRS1, phosphorylated insulin receptor substrate 1; pAKT, phosphorylated AKT; GAPDH, glyceraldehyde 3 phosphate dehydrogenase. a,b,c,dValues within groups indicate significant difference at P<0.05.
|
Godex ameliorated β-oxidation and gluconeogenesis through a CrAT-mediated pathway in Hepa-1c1c cells
CrAT siRNA diminished the effect of Godex on CrAT mRNA and protein expressions (Fig. 6A and B), whereas increased SREBP-1 expression (Fig. 6C) in Hepa-1c1c cells. The Godex-induced increases in phosphorylated AMPK, PPARα, phosphorylated ACC, UCP2, and CPT-1 were reversed by treatment with CrAT siRNA. The gluconeogenic enzymes PEPCK and G6Pase were also down-regulated by Godex, which were reversed by CrAT siRNA. The Godex-induced increases in CPT-1 (Fig. 6D), CPT-2 (Fig. 6E), CACT, and CrAT (Fig. 6F) protein expressions were reversed by treatment with CPT-1/2, and CACT siRNAs, respectively.
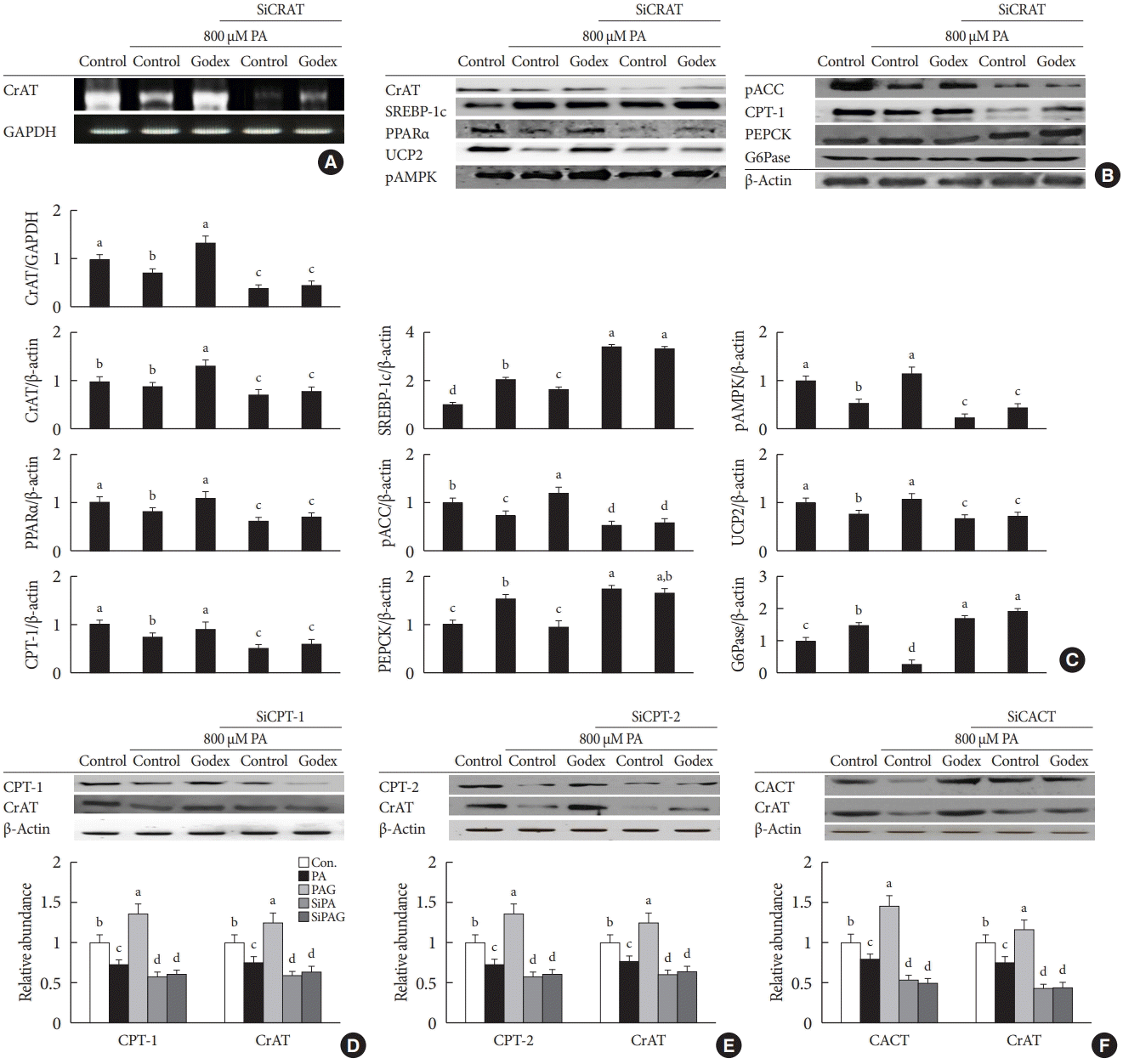 | Fig. 6.Carnitine orotate complex (Godex) ameliorated β-oxidation and gluconeogenesis through a carnitine acetyltransferase (CrAT)-mediated pathway in Hepa-1c1c7 cells. Hepa-1c1c7 cells were transfected with CrAT siRNA and then challenged with 800 μM palmitic acid (PA) with 150 μg/mL Godex. The effectiveness of siRNA knockdown was determined by measuring the expression of CrAT using (A) reverse transcription polymerase chain reaction (RT-PCR), (B) Western blotting, (C) CrAT mRNA level, mitochondrial CrAT protein level, sterol regulatory element-binding protein-1 (SREBP-1), phosphorylated 5’AMP-activated protein kinase (pAMPK), peroxisome proliferator-activated receptor alpha (PPARα), phosphorylated acetyl-CoA carboxylase (pACC), uncoupling protein 2 (UCP2), carnitine palmitoyltransferase-1 (CPT-1), phosphoenolpyruvate carboxykinase (PEPCK), and glucose 6-phosphatase (G6Pase) were assessed by Western blotting. Godex ameliorated CrAT through a carnitine metabolismmediated pathway in Hepa-1c1c cells. Hepa-1c1c7 cells were transfected with CPT1/2 and carnitine acylcarnitine translocase (CACT) siRNA and then challenged with 800 μM PA with 150 μg/mL Godex. The effectiveness of siRNA knockdown was determined by measuring the expression of (D) CPT-1 and CrAT, (E) CPT-2 and CrAT, (F) CACT and CrAT using Western blotting. Data are presented as mean±standard deviation (n=10). GAPDH, glyceraldehyde 3 phosphate dehydrogenase. a,b,c,dValues within groups indicate significant difference at P<0.05.
|
Go to :
DISCUSSION
Impaired glucose and/or lipid metabolism, a sign of obesity and T2DM [6], increases hepatic gluconeogenesis and inhibits the peripheral utilization of glucose, which eventually could lead to insulin resistance [19]. As to the components of Godex, it was reported that pyridoxine (vitamin B6) could help in reactive oxygen species scavenging [20] and improved the insulin resistance [21]. Riboflavin is a critical component of the electron transporters and involved in β-oxidation, improves insulin resistance and gluconeogenesis, and protects from cellular oxidative injury [22]. Biphenyl dimethyl dicarboxylate, which has been reported to significantly lower elevated serum transaminase levels in hepatotoxicity animal mode [23]. Liver extract is rich in vitamin B12, which makes up the DNA. Adenine is a cofactor of nicotinamide adenine dinucleotide (NAD), and signaling molecules such as cyclic adenosine monophosphate (cAMP). The carnitine comprises 51% of Godex. Therefore, the content of carnitine included in G2000 is the amount corresponding to 500 mg/kg of carnitine.
Despite Godex may play a potential role in glucose metabolism and β-oxidation in fatty liver disease [5-9], the specific role of Godex in NAFLD, which is closely related to hepatic steatosis and insulin resistance, has not been elucidated. G2000 treatment lead to higher RQ levels, indicating that they mostly oxidized carbohydrates despite HFD feeding, which could prevent hyperglycemia. Although we did not perform detailed studies on glucose oxidation, there have already been reports which showed significant relationship between carnitine (main component of Godex), and glycolysis [24-29]. The remarkable finding about Godex treatment in our study is that despite substantial increase in the expression of fatty acid oxidation genes, glucose oxidation was remarkably increased as well, even in the HFD-fed group. It has been reported that decreased level of hexokinase (HK) in streptozotocin (STZ)-induced diabetic rodents inhibits glucose conversion into glucose-6-phosphate leading to hyperglycemia [24]. Up-regulations of hepatic HK expression [25] and muscle phosphofructokinase (PFK) of the obese rats by carnitine treatment were already demonstrated, which suggest the ability of carnitine to increase hepatic glycolysis [25] and may explain the mechanism through which they exert their roles in glucose homeostasis [26]. Intriguingly, Godex-treated mice did not show differences in lipid oxidation and energy expenditure compared with HFD mice and no significant change was observed in browning markers such as UCP1 and CD137 in adipose tissue despite changes in body weight and visceral fat volume.
Fatty acid metabolism has been implicated in mediating the severity of insulin resistance. In the insulin resistant states, fatty acids are favored as an energy source over glucose, which is thus associated with increased fatty acid oxidation, and an overall decrease in glycolysis and glucose oxidation [30]. In animal studies, mice fed a HFD showed insulin resistance in which the accumulation of diacylglycerol has been implicated, likely involving parallel signaling pathways [31]. A HFD also results in accumulation of fatty acid oxidation byproducts in muscle, further contributing to insulin resistance [30]. CrAT has an essential role because of its need for large amounts of carnitine. Carnitine supplementation resulted in a more pronounced switch between fat oxidation and carbohydrate oxidation based on the RQ data [27] and a significant increase in carbohydrate oxidation rate [28] due to the presence of cytosolic CrAT and reverse CrAT activity [30]. Mingrone et al. [29] found that carnitine administration increased glucose oxidation in the T2DM patients. Godex is expected to increase HK and PFK expressions due to the effect of carnitine, and we think that there’s a possibility of the doses of carnitine in our experiment, which we are going to evaluate in our next study using higher doses of carnitine rather than Godex. Considering that our experimental animal is a diet-induced obesity model, Godex has been proven to inhibit the development of obesity as well as switch from fatty acid oxidation to glucose oxidation, which is associated with the improvement of insulin resistance.
SREBP-1c expression was significantly downregulated in Godex-treated groups in a dose-dependent manner, which suggests an improvement in hepatic steatosis. AMPK downregulates hepatic SREBP-1c and thereby prevents the feed-forward transcription of the mRNA level [32]. Godex treatment significantly increased the phosphorylation of AMPK and its direct substrate ACC. CPT-1 activity in the liver was increased by reducing the malonyl-CoA content.
In the HFD group not only the hepatic cholesterol content but also the serum LDL-C content was increased. However, Godex decreased hepatic cholesterol accumulation and hypercholesterolemia through decreases in HMGCR, SREBP2, PCSK9, and LDLr expression.
SREBP2 regulates genes involved in cholesterol synthesis and uptake, including HMGCR, LDLr, and Pcsk9 [33]. Mechanistic studies determined that PCSK9 binds to the LDLr and promotes its lysosomal degradation, in the liver [34,35]. Deletion of SREBP2 in the liver reduced the amount of LDLr mRNA by approximately 20% but there was an accompanying approximately 80% reduction in the mRNA level of PCSK9. PCSK9 is a secreted protein that degrades LDLr in liver [34]. In livers of hepatocyte-Srebf2−/− mice, the reduction in LDLr production was balanced by the reduction in PCSK9-mediated LDLr destruction [36]. The Godex-mediated downregulation of SREBP2 may accompany the downregulation of the LDLr. These results suggest that Godex could regulate not only hepatic fat accumulation by increasing fatty acid oxidation and reducing hepatic lipogenesis but also cholesterol homeostasis.
In hepatocytes, acetyl-CoA is used as a substrate for ketogenesis [37] during extended fasting or limited carbohydrate availability, and the liver adapts to dwindling glycogen stores by increasing β-oxidation and by converting the acetyl-CoA to ketone bodies, primarily acetoacetate and its chemically reduced analog β-hydroxybutyrate [38]. G2000 treatment significantly decreased β-hydroxybutyrate than the HFD group, which suggests that Godex acted to reduce hepatic glucose production, primarily by suppressing gluconeogenesis and improved glucose tolerance through the marked reduction of the hepatic lipid contents in HFD mice.
In liver without supplemental Godex, β-oxidation is severely restricted, and fatty acids are preferentially partitioned toward alternative biosynthetic fates [39]. Mitochondrial OXPHOS activities are strongly correlated with insulin sensitivity [40]. HFD significantly decreased the enzymatic activity of OXPHOS complexes with mitochondrial structural damage, but not in Godex-supplemented mice, which suggests that Godex could play a significant role in preventing OXPHOS dysfunction through the increased expressions of PGC1α, NRF1, and SIRT1. Carnitine-mediated inhibition of histone deacetylase 3 may boost PGC1α transcription [41] which is a crucial coactivator for various transcription factors required for mitochondrial biogenesis such as NRF1 and 2 [42]. Phosphorylation of AMPK boosts the ability of PGC1α to promote transcription of its target genes [41] and enhances Sirt1 activity by boosting the NAD+/NADH ratio [43,44].
Multiple PPARα coactivators have been identified to promote β-oxidation in the liver, and PGC1α is a well-characterized PPARα coactivator which promotes β-oxidation [45]. In the fasted state, SIRT1 deacetylates PGC1α and increases its activity [46]. SIRT1 also physically interacts with PPARα and promotes PPARα transcriptional activity in the liver [47]. Godex induced mitochondrial membrane potential, as well as PGC1α, NRF1, and SIRT1, which could increase the capacity for mitochondrial energy production through fatty acid β-oxidation, and improve insulin sensitivity and obesity.
Muoio et al. [10] showed that muscle-specific ablation of CrAT caused systemic glucose intolerance by inhibition of PDH, and blunted the normal postprandial rise in glucose oxidation; however, carnitine treatment, which increases tissue acetylcarnitine efflux, could stimulate the PDH complex in muscle [10,48]. It was previously suggested that CrAT could regulate mitochondrial acetyl-CoA/CoA balance and PDH activity [10,49]. Acetyl-CoA is a critical intracellular metabolite poised to regulate many cellular processes, including transcription, post-translational modifications, and metabolism [50]. G2000 group showed an increase in PDH activity through modulation of CrAT activity and the acetyl-Co/CoA ratio in the liver. Moreover, G2000 group showed a significant increase in PDH activity through modulation of the acetyl-Co/CoA ratio via increased CrAT activity in the skeletal muscle. Significant reduction of plasma lactate level, after the supply of Godex, suggests the activation of PDH, which could increases the oxidation of glucose and OXPHOS from mitochondria. Furthermore, Godex could increase glucose uptake by controlling the expression of membrane GLUT4 in myotubes, which were reversed by blocking CrAT expression.
Although the calorimetry did not demonstrate a significant increase in lipid oxidation, the expression of proteins related to fatty acid oxidation such as PPARα and CPT-1 was significantly increased by Godex, and it is likely that glucose oxidation was more increased as a result of CrAT activation. There’s also a possibility that the lipid oxidation was underestimated by the indirect calorimetry, as well, which should be evaluated in more detail in our future study. Although insignificant, there was a small tendency for lipid oxidation to increase with increasing Godex dose, and when we have administered pharmacological doses of carnitine (not Godex) to the obese animals, we could find significant increases in both lipid and glucose oxidation in association with significant weight loss (unpublished data).
In light of these findings, we hypothesized that CrAT deletion in hepatocytes would amplify metabolic dysregulation. To confirm a causative relationship between CrAT-mediated SREBP1c deactivation and upregulation of β-oxidation, we conducted additional experiments in hepatocytes. Inhibition of the CrAT gene by siRNA effectively abolished β-oxidation and upregulated gluconeogenesis-related protein expression in PA-treated Hepa-1c1c7 cells. Besides, the expressions of proteins of β-oxidation were increased, and gluconeogenesis was decreased in the livers of the HFD group with carnitine, the main component of the Godex, and was reversed by treatment with siCrAT. CrAT knockdown increased HOMA-IR. When siRNAs of CPT-1/2 and CACT were treated, CrAT activity was significantly reduced, which suggests the importance of carnitine metabolism in Godex action.
Our results collectively demonstrate that Godex could improve glucose metabolism by inhibition of hepatic gluconeogenesis and increase in insulin sensitivity in HFD-fed mice by modulating CrAT. However, further mechanistic studies are warranted to clarify the molecular mechanisms.
Go to :
 XML Download
XML Download