

 PDF
PDF Citation
Citation Print
Print



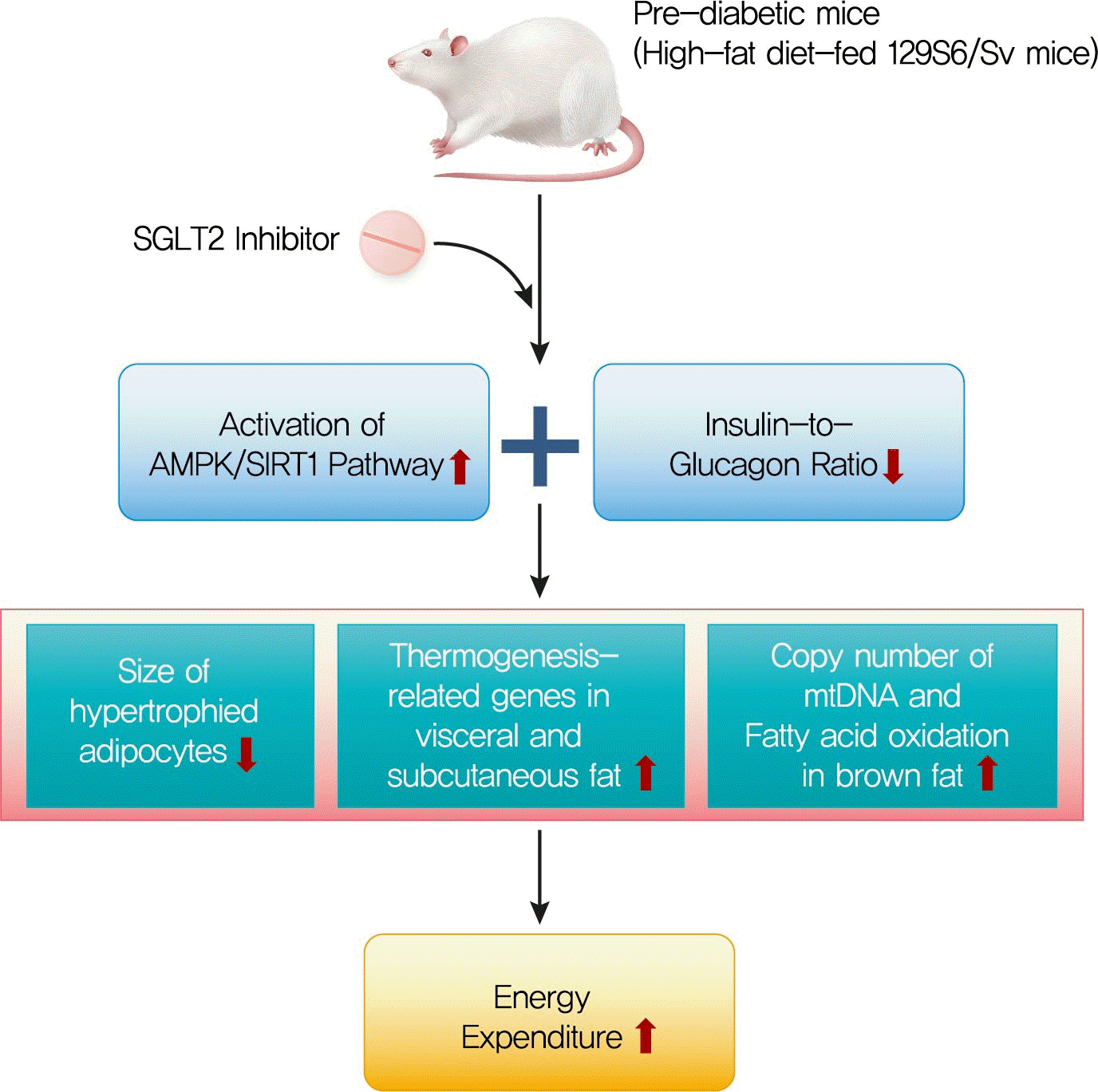
INTRODUCTION
Obesity is a major health problem that is becoming increasingly prevalent; it affects more than one-third of the population worldwide [1]. Recent studies have shown that obesity is closely associated with insulin resistance, inflammatory processes, impaired fatty acid metabolism, and mitochondrial dysfunction [2]. These effects link obesity to many metabolic comorbidities such as type 2 diabetes mellitus, hypertension, hyperlipidemia, non-alcoholic fatty liver disease, and cardiovascular diseases [3].
Adipose tissue consists of two types of adipocytes: white adipose tissue (WAT), which serves as an energy store and has unilocular lipid droplets, and brown adipose tissue (BAT), which has multilocular lipid droplets and a large number mitochondria and highly expresses uncoupling protein 1 (UCP1) in the inner membrane of the mitochondria. BAT dissipates chemical energy as oxidative energy, resulting in the production of heat and an increase in energy expenditure [4,5]. Recently, inducible brown adipocytes, known as “beige adipocytes,” have been studied extensively. The browning of WAT is stimulated by prolonged exposure to cold conditions or treatment with a beta-adrenergic or a peroxisome proliferator-activated receptor γ (PPARγ) agonist and results in high levels of UCP1 [6]. As brown and beige fat increase energy expenditure and have beneficial effects on glucose and lipid metabolism, the manipulation of these pathways has been suggested as a potential treatment for obesity and related metabolic disorders [5].
Sodium-glucose co-transporter 2 (SGLT2) inhibitors are a new class of antidiabetic drugs that were first approved in March 2013 [7]. These agents improve plasma glucose levels via an insulin-independent mechanism that promotes urinary glucose excretion in the renal proximal tubules [8]. SGLT2 inhibitors also exert beneficial effects on body weight and blood pressure by inducing diuresis and natriuresis [9]. In addition, SGLT2 inhibitors are reported to attenuate inflammation and insulin resistance in the adipose tissue and liver of high-fat diet (HFD)-induced obese mice [10]. However, the reports on the effects on energy expenditure and adipose tissue browning are conflicting. A single administration of an SGLT2 inhibitor to nondiabetic mice resulted in the suppression of BAT thermogenesis [11], but chronic inhibition of SGLT2 in HFD-induced obese mice caused enhanced energy expenditure and fat browning [12].
In this study, we examined the effect of chronic administration of an SGLT2 inhibitor (ipragliflozin) on energy metabolism in HFD-fed 129S6/Sv mice. 129S6/Sv mice are resistant to the stimulus of HFD, showing the induction of beige fat with higher browning propensity and less susceptibility to diabetes compared with C57BL/6J mice [13,14]. We chose this model to investigate the effect of ipragliflozin, focusing on adipose tissue browning, irrespective of the glucose-lowering mechanism.
METHODS
Experimental animals and study design
Five-week-old male 129S6/Sv mice were purchased from Taconic (Germantown, NY, USA). After 1 week of acclimatization, the mice were assigned to one of the following three groups: (1) normal chow (NC) diet and treated with vehicle (n=6); (2) HFD (consisting of 60% fat, 20% carbohydrate, and 20% protein, total caloric energy=5.24 kcal/g; D12492; Research Diet Inc., New Brunswick, NJ, USA) and treated with vehicle (n=10); or (3) HFD and treated with ipragliflozin, 10 mg/kg (n=10). The administration dose of ipragliflozin in this study was similar to the animal equivalent dose of clinically used ipragliflozin for human [15]. Each treatment was administered daily by oral gavage for 16 weeks. The animals were housed at a temperature of 23°C±2°C and humidity level of 60%±10% under a 12-hour light/dark cycle and had free access to food and water. All animal procedures were approved by the Animal Care and Use Committee at the Yonsei University College of Medicine (2016-0047) and all experiments were performed in accordance with the relevant guidelines and regulations.
Biochemical measurements
Random glucose concentrations were determined in blood samples obtained via the tail vein. Body weight and food intake were measured weekly over the entire treatment period. An oral glucose tolerance test was performed at week 15 after a 9-hour fast using blood samples collected via the tail vein at 0, 15, 30, 60, 90, and 120 minutes after glucose administration (2 g/kg body weight). An insulin tolerance test was performed at week 16 after a 4-hour fast, and blood samples were obtained at 0, 15, 30, 60, and 120 minutes after an intraperitoneal injection of human regular insulin (0.75 units/kg). At the end of the treatment, the animals were euthanized, and blood samples were collected via heart puncture. Serum levels of aspartate aminotransferase, alanine aminotransferase (ALT), triglycerides (TG; Biovision, Milpitas, CA, USA), free fatty acids (Bioassay Systems, Hayward, CA, USA), insulin (Alpco, Salem, NH, USA), glucagon (Biovision), and β-hydroxybutyrate (Abcam, Cambridge, UK) were measured. Hepatic TG levels were determined using the triglyceride quantification kit (K622; Biovision) according to the manufacturer’s instructions.
Histological and immunohistochemical analyses
Paraffin-embedded liver and adipose tissue sections were stained with hematoxylin and eosin. For staining of lipids, frozen sections of the liver were stained with Oil Red O. Sections of visceral and subcutaneous WAT and BAT were immunohistochemically stained for UCP1 using the anti-UCP1 antibody (ab10983; Abcam). Sections of the pancreas were stained with anti-insulin and anti-glucagon antibodies. Quantitative analyses of the stained area in the pancreas and adipose tissues were conducted using the Image J software (National Institutes of Health, Bethesda, MD, USA).
RNA isolation and real-time polymerase chain reaction analysis
Total RNA was extracted from cells using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Complementary DNA (cDNA) was synthesized using a high capacity cDNA transcription kit (Applied Biosystems, Foster City, CA, USA) and quantitative real-time polymerase chain reaction was conducted on the ABI 7500 sequence detection system using the SYBR Green Master Mix (Applied Biosystems). The levels of acetyl-CoA carboxylase (Acc), fatty acid synthase (Fas), Cd11c, monocyte chemoattractant protein 1 (Mcp1), Ucp1, type 2 selenodeiodinase (Dio2), Cidea, peroxisome proliferator-activated γ coactivator 1α (Pgc1α), transmembrane protein 26 (Tmem26), sirtuin 1 (Sirt1), cytochrome c oxidase subunit II (Cox2), carnitine palmitoyltransferase 1a (Cpt1a), acyl-coenzyme A dehydrogenase (Acad), and acyl-CoA synthetase long-chain family member 1 (Acsl1) messenger RNAs (mRNAs) were determined using specific primers, the used sequences of which are listed in Supplementary table 1. The expression of target genes was normalized to that of reference gene, glyceraldehyde 3-phosphate dehydrogenase (Gapdh). Quantitative analyses were performed using the 2-ΔΔCt method and StepOne Software version 2.2.2 (Life Technologies, Grand Island, NY, USA).
Western blot analysis
Western blot analysis was performed as per the standard protocol. Protein was spectrophotometrically quantified using Bradford reagent. Tissue lysate was mixed with Laemmli buffer (2X, Bio-Rad, Hercules, CA, USA) supplemented with 5% β-mercaptoethanol in a 1:1 ratio. The proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a polyvinylidene difluoride (PVDF) membrane by electrophoretic transfer (110 V for 1 hour). The protein-bound membrane was blocked in a solution of 5% non-fat dried milk prepared in tris-buffered saline with Tween 20 buffer; it was probed overnight with the primary antibody at 4°C and subsequently with the secondary antibody. Specific antibodies against AMP-activated protein kinase (AMPK; #2532S; Cell Signaling, Beverly, MA, USA), pAMPK (#2535S; Cell Signaling), SIRT1 (#28170; Abcam), and GAPDH (#32233; Santa Cruz Biotechnology, Dallas, TX, USA) were used. The chemiluminescent signals were recorded using the SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific, Waltham, MA, USA) in a chemiluminescence imager (Imagequant LAS 4000 mini; GE, Marlborough, MA, USA).
Statistical analyses
Data are expressed as the mean±standard error (SE). Differences between groups were analyzed using one-way analysis of variance (ANOVA) for multiple comparisons and two-way ANOVA for multiple variables. A P<0.05 was considered statistically significant. All statistical analyses were conducted using SPSS for Windows version 23.0 (IBM, Armonk, NY, USA), and the graphs were plotted using GraphPad Prism version 6.0 (GraphPad, San Diego, CA, USA).
RESULTS
Biochemical characterization
During the study period, the weight of the HFD control mice was the highest, whereas that of the ipragliflozin-treated HFD mice was slightly less, but the difference was not statistically significant (Fig. 1A). The daily food intake and random glucose levels were not different between the three groups (Fig. 1B and C). In the oral glucose tolerance test and insulin tolerance test, the areas under the curves did not show any significant difference between the three groups (Fig. 1D and E).
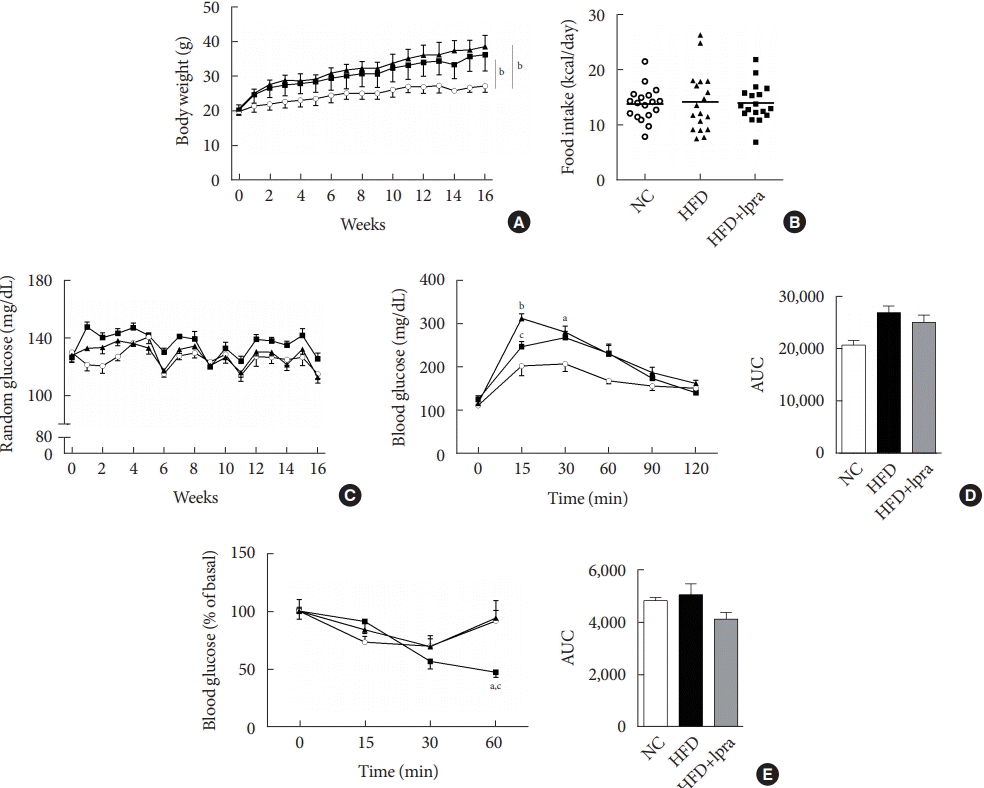
Fig. 1.
Biochemical characterization of the mice. Male 129S6/Sv mice were fed with a normal chow (NC) or high-fat diet (HFD) and were treated with vehicle or ipragliflozin (Ipra) for 16 weeks. (A) Changes in body weight. (B) Food intake (kcal per day). (C) Changes in the random blood glucose levels. (D) Serum glucose levels after an oral glucose tolerance test and area under the curve. (E) Serum glucose levels after an insulin tolerance test and area under the curve. Data are presented as the mean±standard error. NC control (unfilled circle), HFD control (filled triangle), and Ipra-treated HFD mice (filled square). aP<0.05 vs. the NC group, bP<0.01 vs. the NC group, cP<0.05 vs. the HFD group.
![]()
Effects of ipragliflozin on pancreatic islets
There was a slight decrease in the serum insulin level (Fig. 2A), but the serum glucagon level was significantly increased in the ipragliflozin-treated HFD mice (Fig. 2B), resulting in a marked decrease in the insulin-to-glucagon ratio in the ipragliflozin-treated HFD mice (Fig. 2C). There was no difference in the serum β-hydroxybutyrate level (Fig. 2D). Immunohistochemical analysis with anti-insulin antibodies revealed the increased number and size of the pancreatic islets in the HFD control mice, which was improved by ipragliflozin treatment (Fig. 2E). Staining with anti-glucagon antibodies showed that the relative glucagon-positive area was increased by ipragliflozin treatment (Fig. 2F and G), which was consistent with the biochemical findings.
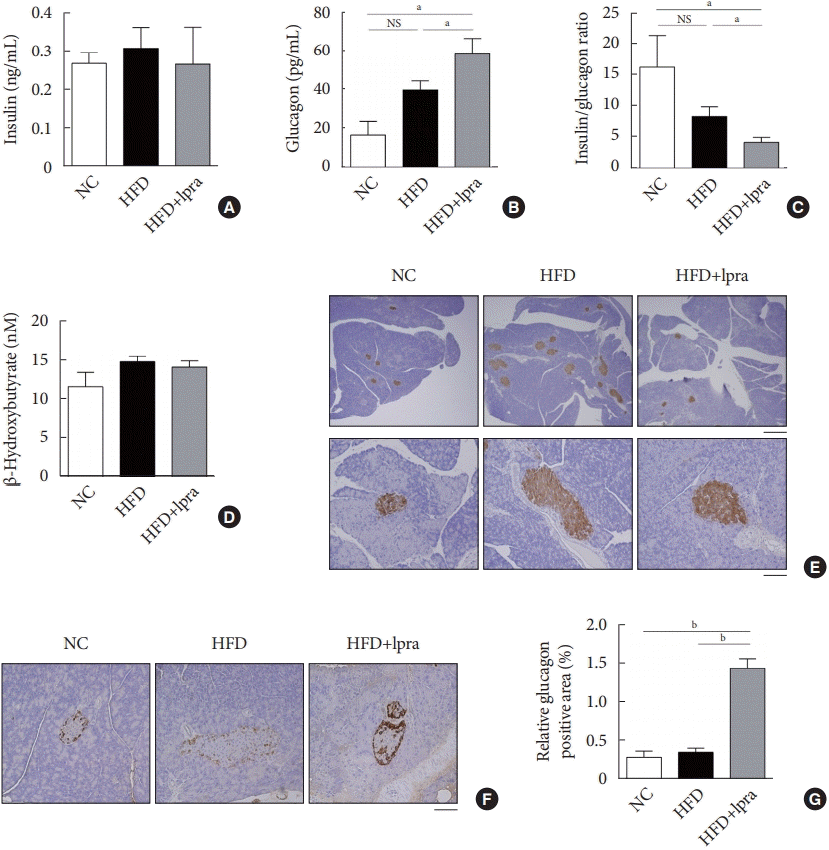
Fig. 2.
Effects of ipragliflozin (Ipra) on pancreatic islets. (A) Serum insulin concentration, (B) serum glucagon concentration, (C) ratio of serum insulin-to-glucagon levels, and (D) serum β-hydroxybutyrate concentration. (E) Immunohistochemical analysis of insulin using pancreatic tissue sections. Scale bar, 500 μm (upper) and 100 μm (lower). (F) Immunohistochemical analysis of glucagon using pancreatic tissue sections. Magnification, ×200. (G) Relative glucagon-positive area (%) in a pancreatic section. Data are presented as the mean±standard error. NC, normal chow; HFD, high-fat diet; NS, not statistically significant. aP<0.05, bP<0.01.
![]()
Ipragliflozin ameliorates HFD-induced hepatic steatosis
We evaluated the effects of ipragliflozin on the liver after 16 weeks of treatment. The weight of the liver was slightly lower and the serum levels of ALT and TGs tended to decrease in the ipragliflozin-treated HFD mice group compared with the values in the HFD control mice (Fig. 3A-D). Histological analyses of the liver showed severe hepatic steatosis in the HFD control mice, which was markedly attenuated by ipragliflozin (Fig. 3E and F). The hepatic TG content was much lower in the ipragliflozin-treated HFD mice than in the HFD control mice (Fig. 3G). These effects were associated with the suppression of lipogenesis-related and proinflammatory genes (Fig. 3H and I) as well as the increased phosphorylation of AMPK and SIRT1 (Fig. 3J), suggesting that ipragliflozin ameliorated HFD-induced hepatic steatosis via the activation of the AMPK/SIRT1 pathway.
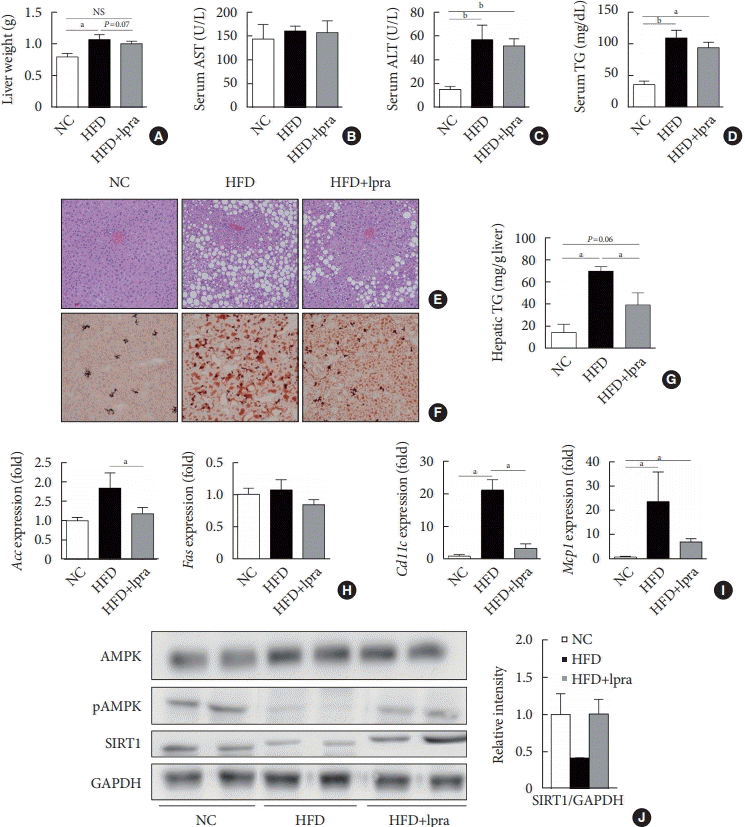
Fig. 3.
Effects of ipragliflozin (Ipra) on liver panels and hepatic steatosis. (A) Liver weight. (B, C, D) Serum aspartate aminotransferase (AST), alanine aminotransferase (ALT), and triglyceride (TG) levels. (E) Hematoxylin and eosin (H&E) staining and (F) Oil Red O staining of liver sections. Magnification, ×200. (G) Hepatic TG concentration. (H, I) Relative mRNA expression levels of lipogenesis-related and proinflammatory genes. (J) Protein levels of AMP-activated protein kinase (AMPK), pAMPK, and sirtuin 1 (SIRT1) in the liver were determined using Western blot analysis. The graph on the right shows the densitometric analysis of the SIRT1/glyceraldehyde 3-phosphate dehydrogenase (GAPDH) ratio determined from the immunoblots shown on the left. Left, normal chow (NC) control mice. Middle, high-fat diet (HFD) control mice. Right, Ipra-treated HFD mice. Data are presented as the mean±standard error. NS, not statistically significant; Acc, acetyl-CoA carboxylase; Fas, fatty acid synthase; Mcp1, monocyte chemoattractant protein 1. aP<0.05, bP<0.01.
![]()
Ipragliflozin treatment reduces the mass of white adipose tissue and the size of adipocytes
We investigated whether ipragliflozin has a beneficial effect on WAT remodeling. The weights of the visceral and subcutaneous fat were slightly decreased by ipragliflozin treatment, even though the differences were not statistically significant (Fig. 4A and B). The administration of ipragliflozin markedly reduced the size of the adipocytes as evident in the histological analyses (Fig. 4C). The average size of the adipocytes was significantly decreased and was equal to or less than that in the NC control mice, suggesting that ipragliflozin ameliorated the adipocyte hypertrophy caused by the HFD, resulting in smaller adipocytes (Fig. 4D and E).
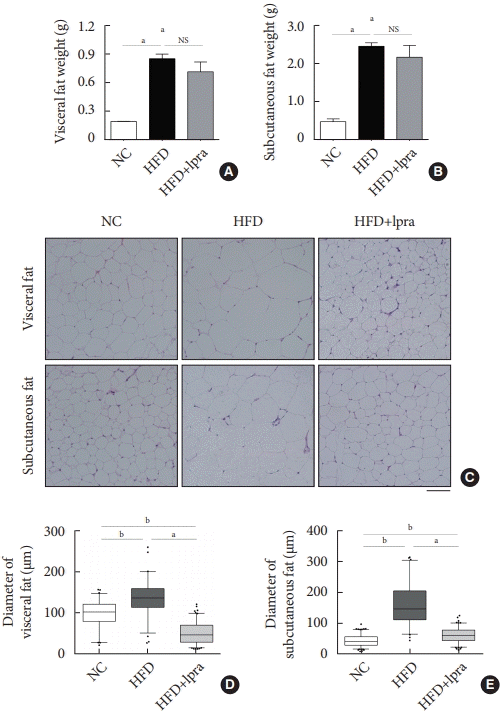
Fig. 4.
Effects of ipragliflozin (Ipra) on the mass of white adipose tissue and size of adipocytes. (A) Visceral fat weight and (B) subcutaneous fat weight. Data are presented as the mean±standard error. (C) Hematoxylin and eosin (H&E) staining of visceral and subcutaneous fat. Scale bar, 100 μm. (D, E) Histogram of visceral and subcutaneous fat diameters. The median is marked by a vertical line inside the box, the ends of the box are the upper and lower quartiles, and the lower and the upper lines outside the box represent the 5th and 95th percentiles, respectively. Whisker plots represent the outliers. NC, normal chow; HFD, high-fat diet; NS, not statistically significant. aP<0.05, bP<0.01.
![]()
Ipragliflozin induces brown fat-like changes in white adipose tissue
Next, we examined whether ipragliflozin promotes the browning of WAT. Immunohistochemical analyses of the visceral and subcutaneous WAT showed that the WAT of the HFD mice treated with ipragliflozin contained more UCP1-positive cells than that of the NC or HFD control mice (Fig. 5A). The relative mRNA levels of Ucp1 and other thermogenesis-related genes, including Dio2, Cidea, and Tmem26, were also significantly upregulated by ipragliflozin treatment in both visceral (Fig. 5B) and subcutaneous (Fig. 5D) fat. In addition, the mRNA levels of Sirt1 were significantly increased in both visceral and subcutaneous fat by ipragliflozin treatment (Fig. 5C and E). The decreased phosphorylation of AMPK and SIRT1 in the visceral fat of the HFD control mice was recovered by ipragliflozin treatment (Fig. 5F). These data suggest that ipragliflozin induces brown fat-like changes as well as the activation of the AMPK/SIRT1 pathway.
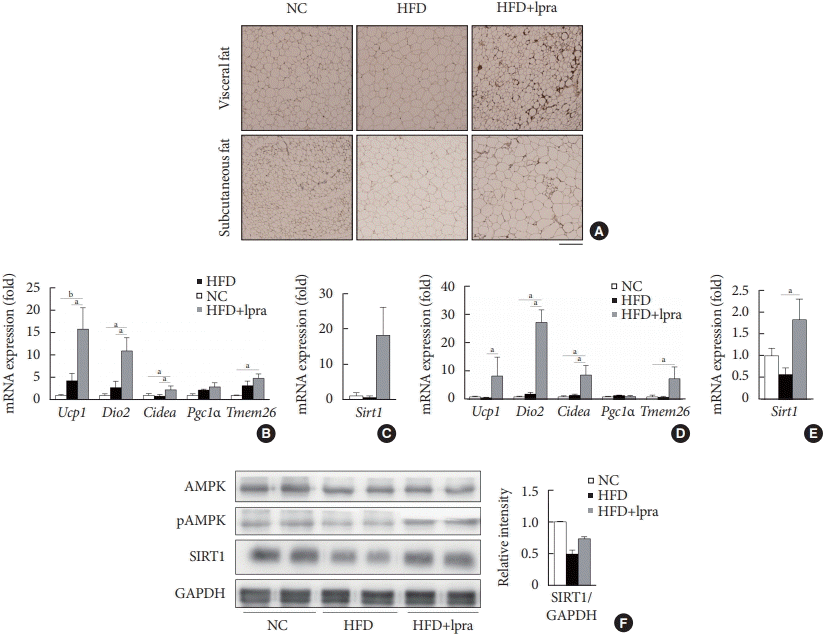
Fig. 5.
Ipragliflozin (Ipra) induced brown fat-like changes in white adipose tissue. (A) Immunohistochemical analysis of the expression of uncoupling protein 1 in the visceral and subcutaneous fat. Scale bar, 200 μm. (B, C) Relative mRNA expression levels of thermogenesis-related genes and sirtuin 1 (SIRT1) in visceral fat and (D, E) subcutaneous fat. (F) Protein levels of AMP-activated protein kinase (AMPK), pAMPK, and SIRT1 in visceral fat were determined using Western blot analysis. The graph on the right shows the densitometric analysis of the SIRT1/glyceraldehyde 3-phosphate dehydrogenase (GAPDH) ratio determined from the immunoblots shown on the left. Left, normal chow (NC) control mice. Middle, high-fat diet (HFD) control mice. Right, Ipra-treated HFD mice. Data are presented as the mean±standard error. Ucp1, uncoupling protein 1; Dio2, type 2 selenodeiodinase; Pgc1α, peroxisome proliferator-activated γ coactivator 1α; Tmem26, transmembrane protein 26. aP<0.05, bP<0.01.e
![]()
Ipragliflozin treatment enhances fat utilization in brown adipose tissue
Compared with that in the NC-fed mice, the gross size and mass of brown fat was increased in the HFD-fed mice (Fig. 6A and B). Although there was no difference in the mass of brown fat upon ipragliflozin treatment, BAT from the HFD control mice contained large lipid droplets, whereas that from the ipragliflozin-treated HFD mice had small multilocular adipocytes similar to those in the NC control mice (Fig. 6C). The relative mRNA levels of Ucp1, Dio2, and Cidea showed no difference upon ipragliflozin treatment in the HFD-fed mice (Fig. 6D). However, the copy number of the mitochondrial DNA was significantly decreased in the HFD control mice; this was markedly restored by ipragliflozin treatment (Fig. 6E). This effect of ipragliflozin was associated with the upregulated mRNA levels of fatty acid oxidation-related genes, including Cpt1a, Acad, and Acsl1 (Fig. 6F).
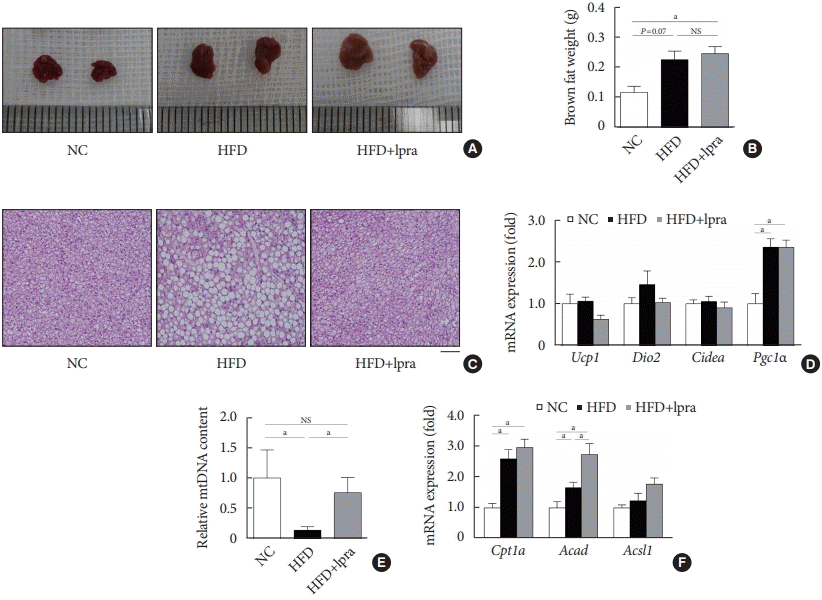
Fig. 6.
Effects of ipragliflozin (Ipra) on brown adipose tissue. (A) Representative gross images of interscapular brown adipose tissue. (B) Brown fat weight. (C) Hematoxylin and eosin (H&E) staining of brown fat. Scale bar, 100 μm. (D) Relative mRNA expression levels of thermogenesis-related genes in the brown fat. (E) Relative mitochondrial DNA content analyzed by polymerase chain reaction using primers specific for cytochrome c oxidase subunit II. (F) Relative mRNA expression levels of fatty acid oxidation-related genes. Data are presented as the mean±standard error. NC, normal chow; HFD, high-fat diet; NS, not statistically significant; Ucp1, uncoupling protein 1; Dio2, type 2 selenodeiodinase; Pgc1α, peroxisome proliferator-activated γ coactivator 1α; Acad, acyl-coenzyme A dehydrogenase; Acsl1, acyl-CoA synthetase long-chain family member 1. aP<0.05.
![]()
DISCUSSION
In this study, we investigated the effects of long-term treatment with ipragliflozin, a selective SGLT2 inhibitor on HFD-induced obese mice. In the 129S6/Sv mouse model, ipragliflozin treatment had no significant effect on glycemic control and weight loss. However, it induced a significant reduction in the insulin-to-glucagon ratio and attenuated HFD-induced hepatic steatosis. The visceral and subcutaneous WAT of the ipragliflozin-treated HFD mice showed an improvement in the hypertrophy of adipocytes as indicated by the presence of small adipocytes. Additionally, ipragliflozin treatment led to the browning of WAT with significantly increased expression of Ucp1, and it promoted fatty acid oxidation and fat utilization in BAT. These effects were found to be independent of the reduction in body weight or the glucose-lowering effects and were accompanied by the activation of the AMPK/SIRT1 pathway.
Among antidiabetic drugs, thiazolidinediones (PPARγ agonists) are well-known key regulators of adipose tissue and promote the browning of WAT by attenuating inflammation in the adipose tissue, improving insulin sensitivity, and upregulating the expression of PRD1-BF-1-RIZ1 homologous domain containing protein-16 (Prdm16) [16,17]. Liraglutide, a glucagon-like protein-1 (GLP-1) receptor agonist, also stimulated BAT thermogenesis and WAT browning in a mouse model [18,19]. Similarly, sitagliptin, a dipeptidyl peptidase-4 inhibitor, enhanced energy expenditure and the expression of Ucp1 in obese mice [20]. However, the effects of SGLT2 inhibitors on energy expenditure and thermogenesis showed conflicting results [11,12]. Long-term SGLT2 inhibition by empagliflozin in HFD-fed diabetic mice, enhanced energy expenditure and thermogenesis and induced M2 polarization in macrophages, leading to adipose tissue browning [12]. In contrast, a single administration of dapagliflozin acutely reduced BAT activity in NC-fed mice by suppressing sympathetic nerve activity and norepinephrine content in BAT [11].
In the present study, we examined the effects of long-term SGLT2 inhibition by ipragliflozin in 129S6/Sv mice. In this model, the mice became obese and developed hepatic steatosis and adipose tissue hypertrophy after being fed with the HFD, but the random glucose levels were in the normal range. These characteristics are similar to those of prediabetic conditions. In a previous study, similar metabolic changes by SGLT2 inhibitor administration were observed in patients with impaired glucose tolerance: increased glucosuria (approximately one-third lower than that in diabetic subjects), decreased insulinto-glucagon ratio, increased endogenous glucose production, and stimulated lipolysis and ketogenesis [21]. However, the effects of SGLT2 inhibition on the beiging of WAT in prediabetic obese mice have not yet been studied. We confirmed that chronic treatment with an SGLT2 inhibitor was associated with the browning of both visceral and subcutaneous WAT and increased fat utilization in interscapular BAT.
The possible mechanisms underlying this phenomenon are as follows: first, the use of SGLT2 inhibitors leads to a reduction in the insulin-to-glucagon ratio. This was reported to be due to a decrease in the plasma insulin levels because of glucosuria [22]. In a recent study, it was proposed that SGLT2 is expressed in pancreatic alpha cells and that SGLT2 inhibition can directly trigger the secretion of glucagon in pancreatic islets [23]. This hormonal change may lead to a decrease in lipogenesis in the liver and an increase in lipolysis in the adipose tissue [24]. Besides regulating glucose and lipid metabolism, glucagon also has a role in the control of energy expenditure and thermogenesis [25]. The administration of glucagon increased the weight of BAT as well as the mitochondrial mass in a rat model [26]. In a previous study using glucagon knockout mice, glucagon was essential for the function of BAT and adaptive thermogenesis via hepatic fibroblast growth factor 21 [27]. Therefore, we suggest that ipragliflozin treatment induced hyperglucagonemia and reduced the insulin-to-glucagon ratio which may have played a role in the browning of adipose tissue.
Second, increased activity of the AMPK/SIRT1 pathway may also play a role. SGLT2 inhibition by cana-gliflozin inhibited the downregulation of SIRT1 expression in the kidney of db/db mice [28]. Here, we found that ipra-gliflozin, an SGLT2 inhibitor, promoted the phosphorylation of AMPK and consequently upregulated SIRT1 in the liver and WAT. As a regulator of energy homeostasis, the concentration of AMPK usually rises when the consumption of energy exceeds its production [29]. AMPK may also activate SIRT1 (which could lead to an increase in gluconeogenesis and fatty acid oxidation in the liver), prevent the accumulation of TG, and increase lipolysis in WAT [30,31]. In addition, SIRT1-mediated PPARγ deacetylation promoted brown features in WAT and increased the expenditure of energy [32]. Exenatide, a GLP-1 receptor agonist, augmented brown remodeling in a Sirt1-dependent manner [19]. Overall, our data suggested that ipragliflozin mediated the browning of adipose tissue, which was promoted by SIRT1 activation.
Our study has some limitations. We did not measure the urinary glucose concentration. In several studies, the administration of an SGLT2 inhibitor to prediabetic animals or humans did not change the normal blood glucose levels but increased the urinary excretion of glucose [33-35]. Therefore, we assume that ipragliflozin might have increased urinary glucose excretion and exerted pharmacological effects in the current study.
In conclusion, we demonstrated that long-term treatment with ipragliflozin has beneficial metabolic effects on HFD-induced obese mice. Ipragliflozin ameliorated hepatic steatosis and improved visceral and subcutaneous adipocyte hypertrophy. Furthermore, ipragliflozin upregulated the expression of Ucp1 and other thermogenesis-related genes, promoted the browning of WAT, and increased the fatty acid oxidation in BAT, independent of the reduction in body weight or the glucose-lowering effects. Based on our results, we could suggest that ipragliflozin, an SGLT2 inhibitor, may have favorable therapeutic potential in prediabetic obese patients.
 XML Download
XML Download