

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
In the past three decades, lipid management has become a mainstay of primary and secondary pharmacological cardiovascular prevention. Many studies have evaluated and validated several cardiovascular risk factors, including age, high low-density lipoprotein cholesterol (LDL-C), low high-density lipoprotein cholesterol (HDL-C), high triglycerides, and inflammation.1) Pharmacological agents have been developed to control these risk factors, and some have shown good efficacy. However, only a few originally developed agents have achieved the final goal of reducing the risk for cardiovascular disease. As mentioned, the most well-known controllable risk factor with clinical benefit is high LDL-C, and the most successful pharmacological approach is lipid-lowering, particularly with statins.2)
Since around 2010, genetic studies using new techniques began to report novel associations among human genes, intermediate phenotypes such as lipid profile, and cardiovascular risk. Interestingly, a considerable proportion of vascular disease-associated genetic signals is linked to biological pathways relevant to traditional risk factors.3) These findings indicate that diagnosis and treatment of patients with dyslipidemia and as well as investigation of new risk factors and treatment targets are critical issues.
Here, I present the role of genetics in care of patients with dyslipidemia and in cardiovascular disease prevention. Particularly, its role in diagnosis, personalized treatment, and drug discovery and development is discussed.
Go to : 
GENETICS FOR DIAGNOSIS OF FAMILIAL HYPERCHOLESTEROLEMIA
Familial hypercholesterolemia (FH) is the most frequent monogenic autosomal dominant disorder that causes premature coronary artery disease. Patients with FH are generally diagnosed by clinical criteria after active suspicion by physicians.4)5) Several clinical criteria including Dutch, Simon Broome, and Make Early Diagnosis to Prevent Early Death criteria are being used. Diagnosis by such criteria is based on cholesterol levels, physical findings, history of vascular disease, and family history of hypercholesterolemia or premature coronary artery disease. However, there are limitations in clinical diagnosis of FH because it is frequently difficult to obtain sufficient data essential for FH diagnosis from patients. When we encounter a patient with vascular disease who is receiving lipid-lowering therapy (LLT), it can be hard to assess their lipid profile without pharmacotherapy after sufficient wash-out of medications. It can be difficult or impossible to get exact information regarding physical findings, onset age of coronary artery disease, or pre-treatment cholesterol levels of family members. In this regard, genetic testing is not only useful, but also essential for diagnosis in some patients suspicious of FH.
Genetic screening and interpretation of FH testing can be performed in several ways, as depicted in Figure 1. One method is serial single-gene testing (sequential gene testing, that evaluates LDLR, the most common causative gene in FH, and checks for any deletion or duplication. Thereafter, APOB and PCSK9 are sequenced. However, after cost-reduction in testing such as next-generation sequencing, it is possible to screen relevant mutations with lower cost and higher efficiency. LDLR, APOB, PCSK9, and other lipid-associated genes can be included in a single panel (multi-gene testing). Such sequencing can uncover: known pathogenic mutations, novel mutations, or no monogenic pathogenic mutations. After identifying a novel mutation, it is necessary to determine its functionality. Methods for this are familial cosegregation, in silico analysis, and in vitro experiment. In an example of cosegregation in a family with FH,6, the proband (red arrow), a 48-year-old man, showed a D834Rfs/- heterozygous mutation (Figure 2A and B). A sister of the proband had an affected phenotype with the same mutation, whereas his 2 children's cholesterol levels were not elevated, and they did not have the mutation (Figure 2A and B). Conversely, diverse public genetic data can help provide an appropriate decision about the pathogenicity of a mutation, even when its functionality has not been proven.
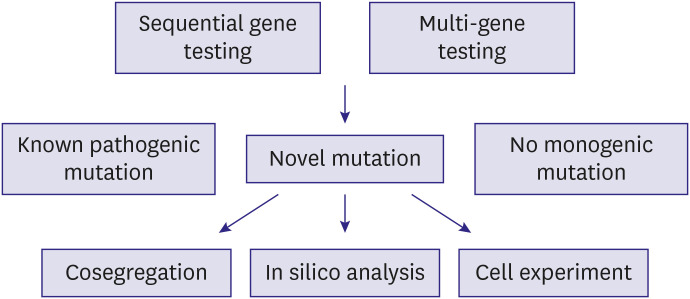 | Figure 1Flow of genetic testing for diagnosis of familial hypercholesterolemia. Any of the sequential or multi-gene testing methods can be used to identify pathogenic mutations. When novel mutations are found, their functionality can be examined through familial cosegregation, in silico analysis, or in vitro experiment assessing the function of low-density lipoprotein uptake.
|
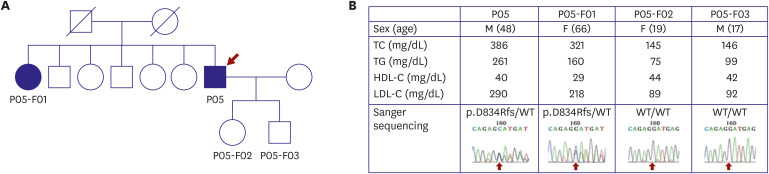 | Figure 2Example of familial cosegregation of a potential pathogenic mutation. (A) Pedigree shows people whose DNA was examined. The proband (P05), proband's sister (P05-F01), proband's daughter (P05-F02), and proband's son (P05-F03) were included. (B) The cholesterol levels of the proband and his sister were compatible with FH, whereas his 2 children had normal levels. Sanger sequencing revealed the same heterozygous mutation in the proband and his sister, whereas no mutations were present in his children (from Han et al.6)). The study including this figure was approved by the Institutional Review Board of Severance Hospital, Seoul, Korea (No. 4-2008-0267) and the participants gave written informed consent.
|
Go to :
GENETICS FOR PATIENTS WITH OTHER EXTREME LIPID PHENOTYPES
Screening of genetic variants and proteins associated with lipid metabolism in patients with extreme lipid phenotypes has been used to discover targets influencing clinical outcomes.7) In previous studies, we analyzed rare and common genetic variants in Koreans with extreme lipid phenotypes. After screening more than 10,000 patients, 22 whose LDL-C levels without LLT were below the first percentile (48 mg/dL) of the Korean population were analyzed. Two target genes, APOB and PCSK9, which are major contributors to the hypobetalipoproteinemia phenotype, were sequenced. In this study, rare variants of APOB or PCSK9 were identified in nine subjects, while the others had common variants of the 2 genes (Figure 3A).8)
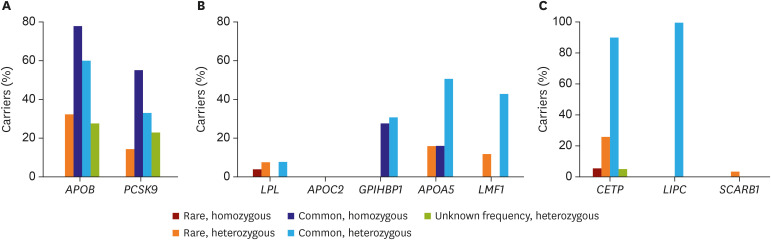 | Figure 3The prevalence of rare and common variant carriers in populations with 3 extreme lipid phenotypes. (A) Carriers of APOB or PCSK9 variants in people with very low low-density lipoprotein cholesterol level (from Lee et al.8)). (B) Carriers of LPL, APOC2, GPIHBP1, APOA5, or LMF1 variants in people with very high triglyceride levels (from Lee et al.).9) (C) Carriers of CETP, LIPC, or SCARB1 variant in people with very high high-density lipoprotein cholesterol levels (from Lee et al.).10)
|
In another study, 26 patients with very high triglyceride level (885 mg/dL [10 mmol/L]) were selected. Five candidate genes, LPL, APOC2, GPIHBP1, APOA5, and LMF1, relevant to triglyceride lipolysis were sequenced. In the analysis, rare homozygous variants of the five genes were very uncommon, while rare heterozygous variants were frequently identified (Figure 3B).9) Conversely, 42 subjects with HDL-C level greater than 100 mg/dL were analyzed for three target genes, CETP, LIPC, and SCARB1. Two rare variants of CETP were found in 13 patients, and one rare variant of SCARB1 was found in a single subject. All subjects had at least one of four common CETP or LIPC variants (Figure 3C).10)
Testing result for monogenic and pathogenic variants in individuals with an extreme lipid phenotype is often negative. Most cases in this population are thought to be manifesting the combined effects of rare heterozygous and common variants of lipid-related genes. Understanding the effects of genetic variants associated with extreme lipid phenotype can provide insight into its potential therapeutic targets as well as its pathophysiology.
Go to :
GENETICS FOR PERSONALIZED TREATMENT OF DYSLIPIDEMIA AND CARDIOVASCULAR PREVENTION
As the therapeutic strategy for patients with FH and pathogenic mutations is the same as for those without mutations, many physicians are skeptical about performing genetic tests in individuals suspicious of FH. However, genetic information is helpful for determining which FH patient is at higher risk for cardiovascular disease and thus who needs more aggressive LLT. In a Japanese study, patients with the FH phenotype and pathogenic monogenic mutations showed about three times higher cardiovascular risk than those without mutations.11)
In addition, knowledge regarding an individual's genetic characteristics can allow prediction of patients with poor response to LLT. Patients with FH and null mutations have shown higher cholesterol levels and lower rates of target achievement.12)13) In a large Spanish study, the presence of defective mutations was a predictor of the ability to reach goal cholesterol levels.14) In our previous study, patients without pathogenic mutations showed better achievement in LDL-C reduction following a statin/ezetimibe-based regimen.15) Furthermore, the lipid-lowering response to a PCSK9 inhibitor was numerically higher in individuals with defective mutations compared to those with null mutations. Interestingly, this finding was also observed in patients with homozygous FH (Figure 4).15)
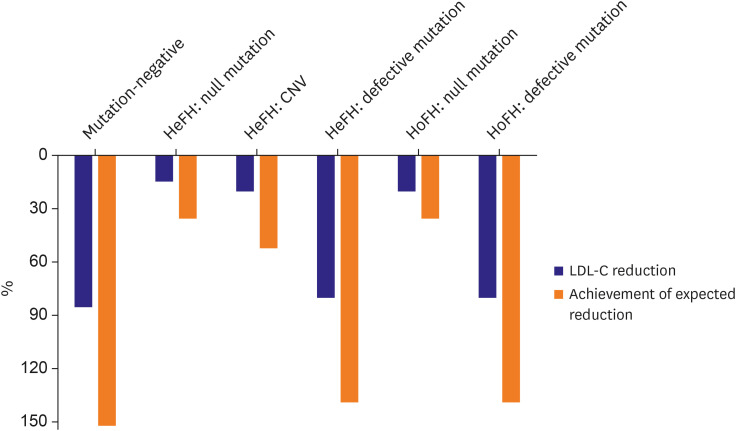 | Figure 4Achievement of expected LDL-C reduction through evolocumab treatment in patients with FH. Blue bars indicate LDL-C reduction following addition of evolocumab to the statin/ezetimibe regimen. Orange bars indicate the achievement of expected LDL-C reduction (adjusted lipid-lowering response) following addition of evolocumab to the statin/ezetimibe regimen. Differences in LDL-C reduction and adjusted response are observed according to mutation type, irrespective of disease homozygosity (From Kim et al.15)).CNV = copy number variation; FH = familial hypercholesterolemia; HeFH = heterozygous familial hypercholesterolemia; HoFH = homozygous familial hypercholesterolemia; LDL-C = low-density lipoprotein cholesterol.
|
Researchers have tried to identify specific genes and their variants to predict response to LLT and to use these results in personalized therapeutic approaches. Recent high-throughput studies have identified genetic loci that are associated with response to LLT. A metagenome-wide association study using more than 18,000 patients confirmed that four loci close to SORT1/CLESR2/PSRC1, LPA, SLCO1B1, and APOE were linked to the response to statins.16) A study coined a genetic score based on 57 SNPs associated with coronary artery disease and showed that it was correlated with clinical benefit of statins.17) Although data including those of the above-mentioned studies are not sufficient for active clinical use, further research on this issue can suggest a more refined clue to individualized LLT. Taken together, information regarding an individual's genetic characteristics is helpful for selection of patients that require more active treatment, prediction of drug response, and choice for more appropriate LLT regimen.
Go to :
GENETICS FOR DRUG TARGET DISCOVERY AND DEVELOPMENT
The progression of technologies related to genomic research and collected data in the past decade have imparted opportunity to discover drug targets. For researchers, high throughput sequencing and genome-wide association studies have been available since the late 2000s; they have continuously gotten less expensive since around 2010. At the same time, great advances in bioinformatic analysis techniques have provided tools to identify genes and variants positively or negatively associated with extreme lipid traits and cardiovascular risk.18) Analysis of large populations with the above-mentioned techniques made it possible to uncover unbiased, causal relationships among lipid levels, cardiovascular phenotypes, and rare genetic variants, via so-called “Mendelian randomization study.”19)20)21) This kind of work was not possible in older studies that used target approaches or simple association analyses realized by traditional techniques and relatively small budgets. These new-generation studies in the field of dyslipidemia and cardiovascular prevention report on the genes related to lipolysis and their clinical impact (Figure 5).22) Studies investigating the relationship between hypotriglyceridemia or combined hypolipidemia and ANGPTL3,23)
APOC3,19) or ANGPTL4
24) (all linked to lipoprotein lipase activity) have shed light on development of therapeutics targeting these genes or gene-related products.
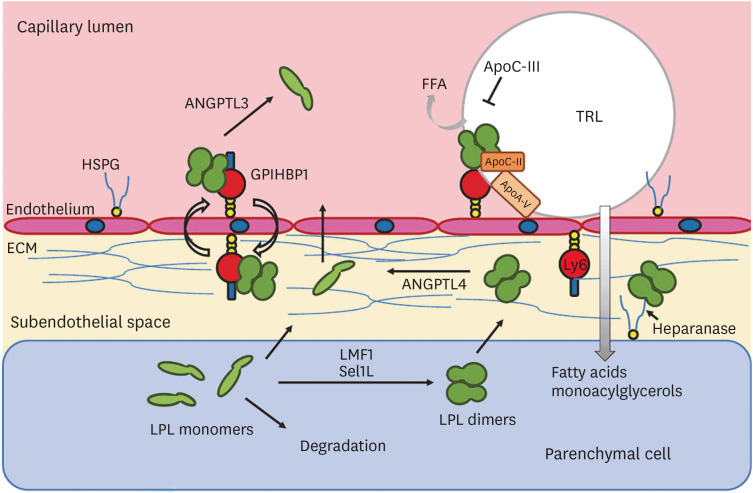 | Figure 5LPL and other related proteins supporting or inhibiting this enzyme acting in endothelial cells (from Olivecrona with permission20)). In the last decade, some of these genes and their proteins have shown considerable influence on the risk of coronary artery disease, suggesting a causal effect.ANGPTL = angiopoietin-like protein; ECM = extracellular matrix; FFA = free fatty acid; HSPG = heparan sulfate proteoglycan; GPIHBP1 = glycosylphosphatidylinositol anchored high density lipoprotein binding protein 1; LMF1 = lipase maturation factor 1; LPL = lipoprotein lipase; Sel1L = sel-1 suppressor of lin-12-like 1; TRL = triglyceride-rich lipoprotein.
|
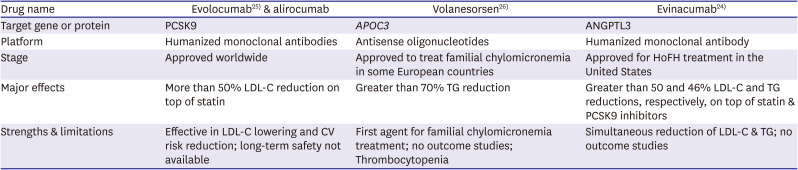
Furthermore, techniques to develop therapeutics to target specific genes, such as antisense oligonucleotides, have become more refined (Figure 6).25) In addition, a few antibody-based therapeutics show promising results in this field26) (Table 1). Collectively, these results demonstrate some powerful pharmacological agents based on genetic knowledge and their clinical impact in the field of lipid management and cardiovascular prevention.27)28)
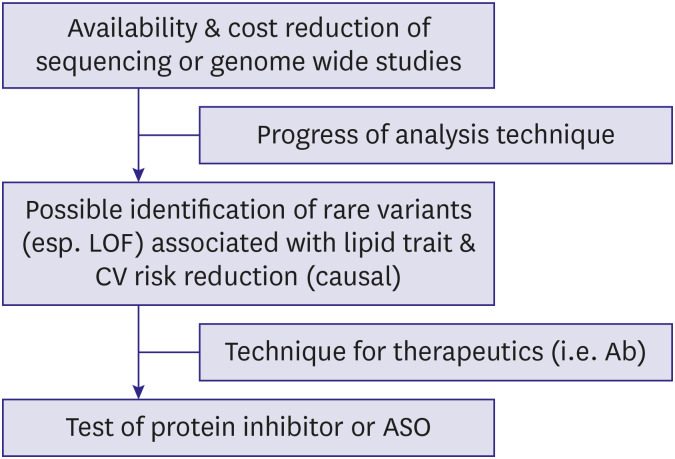 | Figure 6Flow of target discovery and drug development based on recent genetic studies. Progress and cost reduction in genetic analysis and bioinformatics helped the identification of rare functional variants that would have been previously impossible. Improvement in production of gene-targeting therapeutics improved the success rate in drug development.Ab = antibody; ASO = antisense oligonucleotide; CV = cardiovascular; LOF = loss-of-function..
|
Table 1
Summary of recent drug studies based on genetic discovery regarding dyslipidemia
| Drug name | Evolocumab25) & alirocumab | Volanesorsen26) | Evinacumab24) |
|---|---|---|---|
| Target gene or protein | PCSK9 | APOC3 | ANGPTL3 |
| Platform | Humanized monoclonal antibodies | Antisense oligonucleotides | Humanized monoclonal antibody |
| Stage | Approved worldwide | Approved to treat familial chylomicronemia in some European countries | Approved for HoFH treatment in the United States |
| Major effects | More than 50% LDL-C reduction on top of statin | Greater than 70% TG reduction | Greater than 50 and 46% LDL-C and TG reductions, respectively, on top of statin & PCSK9 inhibitors |
| Strengths & limitations | Effective in LDL-C lowering and CV risk reduction; long-term safety not available | First agent for familial chylomicronemia treatment; no outcome studies; Thrombocytopenia | Simultaneous reduction of LDL-C & TG; no outcome studies |
CV = cardiovascular; HoFH = homozygous familial hypercholesterolemia; LDL-C = low-density lipoprotein cholesterol; TG = triglycerides.
![]()
Go to :
CONCLUSION
Genetic testing is useful for diagnosis and risk assessment of patients with FH, a representative monogenic lipid disorder. Genetic studies help us to understand etiology of other extreme lipid phenotypes. Although in its early stage, pharmacogenetic data provide guidance for selection of treatment-eligible patients and optimal lipid-lowering therapeutics. Recent progress in genetic research, bioinformatics analysis, and pharmacological platforms increased the efficiency of discovery of drug targets and development of novel therapeutics.
Ethical statement
The study including Figure 2 was approved by the Institutional Review Board of Severance Hospital, Seoul, Korea (No: 4-2008-0267) and the participants gave written informed consent.
Go to :
 XML Download
XML Download