

 PDF
PDF Citation
Citation Print
Print



Graphical abstract
INTRODUCTION
PATHOLOGY OF NONALCOHOLIC FATTY LIVER DISEASES
Rho-KINASE INHIBITOR AMELIORATES NONALCOHOLIC FATTY LIVER DISEASES
Table 1.
| Animal models with liver diseases | Treatments | Major findings | Reference |
|---|---|---|---|
| Fibrosis induced by CDAA in rats | Y-27632 (1 mg/kg) was treated for 12 weeks | Improvement of hepatic fibrosis and steatosis | [35] |
| Decreased ALT and HA levels | |||
| Reduced hepatic TNFα expression | |||
| Steatosis induced by choline-deficient diet for 6 weeks in rats and the liver was subjected to IR injury | Vitamin A-coupled liposomes carrying Y-27632 for targeted HSCs was administered before ischemia induction | Improved the survival rate after IR injury, the liver blood flow, and the portal perfusion pressure | [37] |
| Decreased AST levels | |||
| Steatosis induced by choline-deficient diet for 6 weeks and the liver was s ubjected to IR injury in rats | Fasudil (10 mg/kg) was administered for 30 minutes before ischemia induction | Attenuation of HSCs activation | [36] |
| Improvement of IR injury | |||
| Decreased AST and ALT levels | |||
| Reduced ET-1 serum levels | |||
| Fibrosis induced by CCl4 treatment for 12 weeks in rats | Y-27635 (30 mg/kg) was treated for 6 weeks | Prevented the development of liver fibrosis | [46,50] |
| Decreased αSMA and TGFβ1-positive cell | |||
| Decreased total collagen | |||
| Increased survival rate after hepatectomy | |||
| CCl4-induced acute liver injury in mice | Y-27632 (1.5 mg/kg) was injected at 24 and 48 hours after CCl4 injection | Reduction of local activation of HSCs | [47] |
| Decreased αSMA | |||
| CCl4-induced acute liver injury in mice | Single injection of HA-1077 (10 mg/kg) | Reduced hepatic apoptosis | [51] |
| Decreased ALT levels | |||
| Fibrosis induced by CCl4 treatment for 7 weeks in mice | Y27632 conjugate (45 mg/kg/day) that is taken up in activated HSCs for 2 weeks | Inhibited activation of HSCs | [39] |
| Decreased ALT and AST level | |||
| Reduced liver fibrosis | |||
| Cirrhosis induced by CCl4-intoxication and BDL in rats | Y-27635 coupled with mannose-6- phosphate targeting HSCs activation | Decreased the portal pressure | [38] |
| Decreased the hepatic-portal resistance | |||
| No extrahepatic effects | |||
| Cirrhosis induced by BDL or CCl4 in rats | Y27632 coupled with human serum albumin substituted with PDGFRβ- recognizing peptides (Y27pPBHSA) | Lower portal pressure, hepatic vascular resistance without effect on systemic vascular resistance | [48] |
| Reduced intrahepatic resistance by decreased moesin and MLC | |||
| Mouse model of NAFLD induced by HFD | RKI-1447 (2 and 8 mg/kg) was treated for 3 weeks | Decreased ALT and AST levels | [40] |
| Decreased serum cholesterol and triglyceride levels | |||
| Ameliorated hepatic steatosis | |||
| Diabetic rats made by a combination of HFD and streptozotocin | Fasudil (10 mg/kg) was treated for 14 weeks | Amelioration of liver fibrosis | [41] |
| Inhibited TGFβ1/CTGF pathway and αSMA expression | |||
| Streptozotocin-induced type 1 diabetic rats | Fasudil (2 mg/kg b.i.d., 10 mg/kg b.i.d., H-Fas group) was treated for 8 weeks | Suppression of inflammation and accumulation of the extracellular matrix | [42] |
| Downregulated TGFβ1 and MMP9/TIMP-1 | |||
| Decreased NF-κB activation | |||
| Rat model of DMN-induced fibrosis | Y27632 (30 mg/kg) was orally administered for 4 weeks | Decrease liver fibrosis | [43] |
| Reduced hepatic collagen and hydroxyproline | |||
| Decreased hepatic αSMA expression | |||
| Endotoxin-induced mice made by a combination of LPS and D-galactosamine | Fasudil (40 mg/kg) or Y-27632 (10 mg/kg) by a single injection | Decreased ALT and AST levels | [45] |
| Decreased hepatic apoptosis | |||
| Attenuated LPS-induced liver injury | |||
| Reduced LPS-induced leukocyte adhesion | |||
| Decreased hepatic TNFα and CXC chemokines expression | |||
| LPS-induced acute liver injury in mice | Y-27632 (10 mg/kg) was injected prior to LPS injection | Attenuated LPS-induced liver injury | [44] |
| Reduced the LPS-induced hepatic inflammatory response and oxidative stress | |||
| Protective effects on hepatic mitochondrial function | |||
| Obstructive cholestasis induced by BDL in mice (hepatocellular damage) | Y-27632 (1 and 10 mg/kg) was injected prior to BDL | Decreased serum AST and ALT levels | [52] |
| Reduced CXC chemokines and leukocyte recruitment | |||
| Restored sinusoidal perfusion | |||
| Rats underwent intrahepatic tumor implantation followed by orthotopic liver transplantation | Y-27632 (10 mg/kg) for 28 days | Suppression of cancer cell migration | [53] |
| Suppression of tumor recurrence | |||
| Increased survival rate | |||
| Mice lacking hepatic ROCK1 | HFD for 16 weeks | Prevented HFD-induced fatty liver | [49] |
| Decreased hepatic TG and cholesterol in serum and liver | |||
| Decreased de novo lipogenesis | |||
| Mice overexpressing a constitutively active mutant of ROCK1 in the liver | HFD for 12 weeks | Promoted HFD-induced fatty liver | [49] |
| Increased serum TG and cholesterol levels | |||
| Increased hepatic TG | |||
| Leptin-deficient mice (ob/ob) mice lacking hepatic ROCK1 | No treatment | Prevented the development of fatty liver | [49] |
| Decreased hepatic TG and cholesterol levels |
CDAA, choline-deficient/L-amino acid-defined; ALT, alanine aminotransferase; HA, hyaluronic acid; TNFα, tumor necrosis factor alpha; IR, ischemia-reperfusion; HSC, hepatic stellate cell; AST, aspartate aminotransferase; ET-1, endothelin-1; CCl4, carbon tetrachloride; αSMA, α-smooth muscle actin; TGFβ, transforming growth factor-β; BDL, bile duct ligation; PDGFRβ, platelet-derived growth factor receptor β; MLC, myosin light chain; NAFLD, nonalcoholic fatty liver disease; HFD, high-fat diet; CTGF, connective tissue growth factor; b.i.d., twice a day; MMP9, matrix metallopeptidase 9; TIMP-1, tissue inhibitors of metalloproteinases-1; NF-κB, Nuclear factor kappa B; DMN, dimethylnitrosamine; LPS, lipopolysaccharide; ROCK1, Rho-associated coiled-coil-containing kinase; TG, triglyceride.
![]()
THE IMPACT OF HEPATIC ROCK1 ON OBESITY AND INSULIN SENSITIVITY
CONTROL OF DE NOVO LIPOGENESIS BY ROCK1
Biochemistry and physiology of hepatic de novo lipogenesis
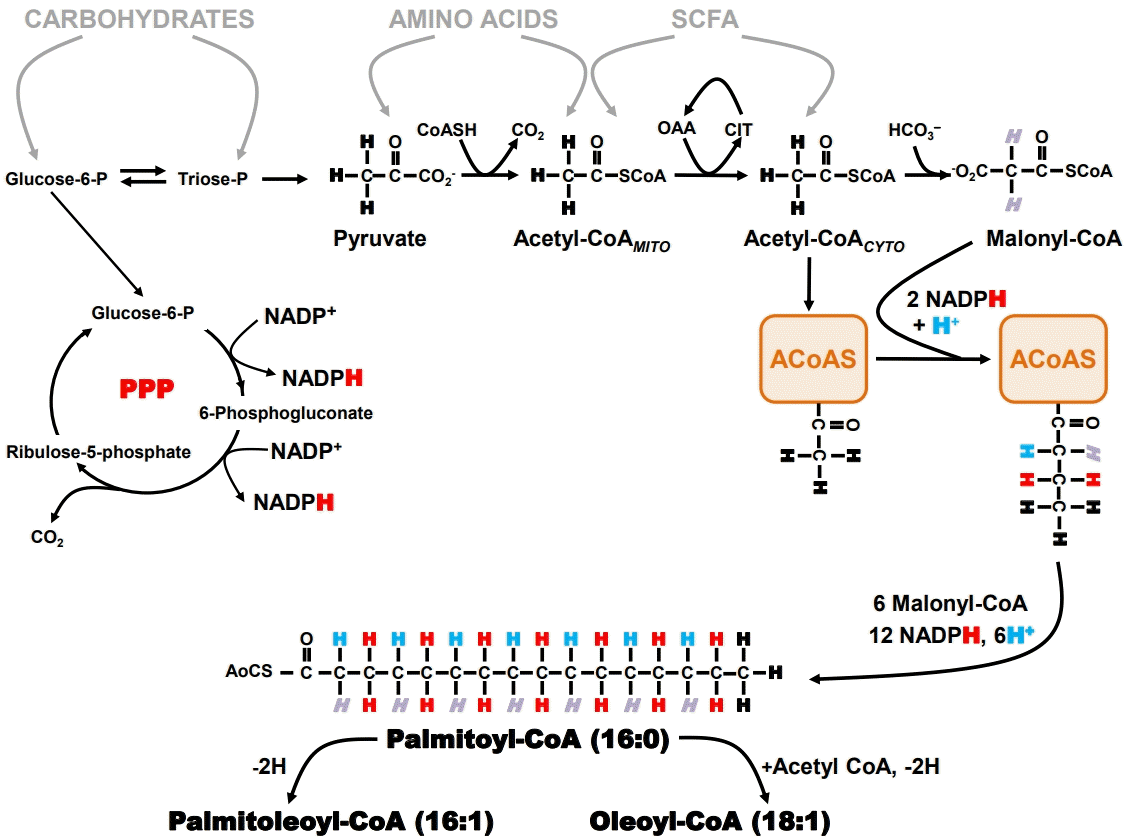 | Fig. 1.Schematic of de novo lipogenesis. Entry points for the main nutrient precursors are shown in gray and the sources of hydrogens of the fatty acyl coenzyme A (acyl-CoA) product are represented by different colors: ‘H+’ hydrogens derived from body water, black ‘H’ for the methyl hydrogens of the initial acetyl-CoA that binds to acyl-CoA synthase, red ‘H’ for the hydrogens derived from nicotinamide adenine dinucleotide phosphate (NADPH) via the pentose phosphate pathway (PPP), and ‘H’ for the hydrogens derived from malonyl-CoA. Also represented is the acyl-CoA synthase complex (ACoAS) following the initial acetylCoA and malonyl-CoA additions. Intracellular acetyl-CoA is represented as distinct mitochondrial (Acetyl-CoAMITO) and cytosolic (acetyl-CoACYTO) pools. CoASH, free acetyl CoA; SCoA, CoA moiety; SCFA, short chain fatty acids; OAA, oxaloacetate; CIT, citrate; AoCS, Co moiety
|
Measurement of hepatic DNL with deuterated water (2H2O)
DNL measurements in animal models and humans
Table 2.
| Study outline | Tracer method for DNL assay | Major findings | Reference |
|---|---|---|---|
| DNL rates were measured in lean healthy subjects and in overweight patients with suspected NAFLD based on transaminase levels and hepatic ultrasound tests. | 2H2O given over 20 hours and enrichment of palmitate within TG-rich lipoprotein measured after a breakfast meal. | This was among the first published study of DNL in NAFLD subjects using the 2H2O tracer. The contribution of DNL to the appearance rate of plasma TG was ~3-fold higher for the NAFLD subjects compared to the healthy controls. | [86] |
| Comparison of DNL contributions to liver TG appearance in obese subjects with low or with high liver fat but matched for other metabolic factors | 2H2O given over 11 days and enrichment of palmitate within TG-rich lipoprotein measured. | Subjects with high liver fat levels had a DNL contribution to liver fat appearance that was twice that of subjects with low liver TG levels. | [84] |
| Determine the sources of fatty acids contributing to hepatic TG and in secreted lipoprotein of NAFLD patients during both fed and fasted states | [1-13C]acetate infused over 4 days. Hepatic TG directly sampled by liver biopsy and plasma lipoprotein TG also sampled. | Analysis of plasma lipoprotein TG yielded similar flux estimates to those of liver TG. DNL accounted for about one-quarter of liver and plasma lipoprotein TG synthesis. DNL was active during both fed and fasted states. | [81] |
| Determine the effect of age-induced skeletal muscle insulin resistance on postprandial hepatic lipid kinetics by studying elderly subjects and young subjects matched for body composition and physical activity | 2H2O given over 2 days and enrichment of palmitate within VLDL measured. | Hepatic DNL rates were two-fold higher in the elderly cohort. Hepatic lipid levels were three-fold higher in the elderly cohort. These were associated with increased fasting plasma glucose levels and increased muscle insulin resistance as seen by impaired muscle glycogen synthesis rates. | [87] |
| Determine the effects of a fatty acid synthase inhibitor (FT-4101) on hepatic DNL rates in NAFLD patients | 2H2O given over 14 days and enrichment of VLDL-palmitate measured during a period of fructose ingestion. | 12 Weeks of FT-4101 administration significantly decreased fractional DNL rates over placebo in a dose-dependent manner. This was accompanied by a significant decrease in liver TG levels. | [88] |
| Compare DNL rates in lean subjects without NAFLD, obese subjects without NAFLD and obese subjects with NAFLD before and after placement on a weight-loss diet | 2H2O given over 3−5 weeks and enrichment of palmitate within TG-rich lipoprotein measured. | DNL rates were highest in obese-NAFLD subjects and were directly correlated with plasma insulin and glucose concentrations. Moderate weight loss resulted in significant decreases in both DNL rates and liver TG levels that was linked to significant decreases in plasma glucose and insulin levels. | [11] |
![]()
Role of ROCK1 in hepatic DNL activity
ROCK1 MEDIATES ENDOCANNABINOID INDUCED LIPOGENESIS
Table 3.
| Animal model of metabolic dysfunction | Treatments | Major findings | Major findings |
|---|---|---|---|
| Obese Zucker fa/fa rats | Rimonabant (30 mg/kg/day) was orally administered for 8 weeks | Decreased plasma ALT, GGT, and ALP levels | [106] |
| Decreased hepatic TNFα levels | |||
| Reduced plasma TG and cholesterol levels | |||
| Improved hepatic steatosis | |||
| OLETF rats | Rimonabant (10 mg/kg/day) was orally administered for 6 weeks | Decreased serum ALT and AST levels | [107] |
| Decreased hepatic TG levels | |||
| Improved hepatic steatosis | |||
| C56BL/6J mice fed a HFD for 20 weeks | Rimonabant (10 mg/kg) was orally administered from 18th to 20th week | Decreased serum AST and ALT levels | [108] |
| Reduced hepatic palmitic, stearic, and oleic acid proportion | |||
| Ameliorated hepatic steatosis | |||
| Sprague-Dawley rats fed a choline-deficient diet for 12 weeks | Rimonabant (10 mg/kg) orally administered from 10th week to 12th week | Decreased serum AST and ALT levels | [109] |
| Decreased hepatic malondialdehyde levels | |||
| Increased hepatic glutathione peroxidase activity | |||
| Reduced hepatic TGFβ immunoreactive area | |||
| Male C57BL/6 mice fed a HFD for 20 weeks | Rimonabant (10 mg/kg) was orally administered from 18th week to 20th week | Decreased serum AST and ALT levels | [110] |
| Decreased adipokines levels (leptin, visfatin, IL-6, IFNγ levels in adipose tissue) | |||
| Reduced hepatic-6, IFNγ levels | |||
| Improved hepatic steatosis | |||
| C57Bl/6J male mice fed a HFD underwent hypoxia exposure for 10 weeks | CB1 receptor agonist (WIN55212-2, 1 mg/kg) was treated for 4 weeks | Hypoxia-induced improvement in hepatic steatosis was abolished by CB1 receptor agonist | [116] |
| Increased hepatic CB1 and SREBP1 expression | |||
| Male Sprague-Dawley and WKY rats injected with LPS/D-galactosamine (acute liver injury model) | CB2 agonist (JWH-133, 2.5 mg/kg) or CB1 antagonist (AM6545, 10 mg/kg) was acutely injected | CB1 antagonist or CB2 agonist had no effect on LPS/D-galactosamine-induced liver injury | [117] |
| NAFLD was induced by a high-fat cholesterol diet for 6 weeks | Cannabis extracts (5 mg/kg) were administrated orally every 3 days for 6 weeks | No change in hepatic lipid accumulation | [118] |
| CBD-rich extracts increased hepatic iNOS and TNFα expression | |||
| THC-rich extracts decreased liver enzymes | |||
| Mice lacking CHOP fed a HFD for 14 weeks | CB1R blocker (JD5037, 3 mg/kg) was administered for 7 days | Failed to reverse DIO-induced reduction of sOB-R levels and hepatic steatosis | [119] |
| Liver fibrosis induced by thioacetamide (200 mg/kg) treatment for 6 weeks in rats | AM1241 (CB2 receptor activator, 3 and 6 mg/kg) was injected for 3 weeks | Suppressed hepatic TNFα, IL-1b, and IL-6 levels | [120] |
| Decreased hepatic TLR4, TGFβ1, αSMA, and miR-155 gene expression | |||
| Inhibited the development of fibrosis | |||
| APOE∗3-Leiden.CETP transgenic mice fed Western-type diet for 20 weeks | Rimonabant (20 mg/kg/day) for 4 weeks | Decreased plasma TG and non-HDL levels | [121] |
| Increased plasma HDL-C levels | |||
| Reduced VLDL-TG production | |||
| Increased VLDL-TG turnover | |||
| Prevented atherosclerosis | |||
| Obese mice fed on a HFD | RTI1092769 (inverse agonist/antagonist of CB1) | Decreased hepatic TG contents | [111] |
| Decreased plasma AST and ALT levels | |||
| Improved hepatic steatosis | |||
| C57BL/6J and fat-1 transgenic mice fed a LFD for 10 weeks +10 weeks of a HFD | Fatty acids SAF oil or LIN oil were supplemented to a LFD for 10 weeks, prior to HFD exposure | Decreased hepatic AEA and 2-AG contents in LIN-treated mice | [122] |
| Improved glucose tolerance and insulin tolerance in LIN-treated mice | |||
| Global CB1 receptor knockout mice | SR161716A (10 mg/kg) was injected before and after CCl4 | Reduced fibrosis associated with chronic liver injury | [123] |
| Decreased hepatic TGFβ1 expression | |||
| Global CB1 receptor knockout mice | No treatment | Decreased hepatic TG levels | [124] |
| Reduced hepatic PLIN2 expression | |||
| Suppressed lipogenesis |
ALT, alanine aminotransferase; GGT, gamma glutamyltransferase; ALP, alkaline phosphatase; TNFα, tumor necrosis factor alpha; TG, triglyceride; OLETF, Otsuka Long-Evans Tokushima Fatty; AST, aspartate aminotransferase; HFD, high-fat diet; TGFβ, transforming growth factor-β; IL-6, interleukin 6; IFNγ, interferon gamma; CB1, cannabinoid receptor type 1; SREBP1, sterol regulatory element-binding protein 1; WKY, Wistar Kyoto; LPS, lipopolysaccharide; CB2, cannabinoid receptor type 2; NAFLD, nonalcoholic fatty liver disease; CBD, cannabidiol; iNOS, inducible nitric oxide synthase; THC, tetrahydrocannabinol; CHOP, C/EBP homologous protein; CB1R, cannabinoid receptor type 1 receptor; DIO, diet-induced obesity; sOb-R, soluble leptin receptor; IL-1b, interleukin 1b; TLR4, toll like receptor 4; αSMA, α-smooth muscle actin; HDL, high-density lipoprotein; HDL-C, high-density lipoprotein cholesterol; VLDL, very low-density lipoprotein; LFD, low-fat diet; SAF, safflower oil; LIN, linseed oil; AEA, arachidonoylethanolamide; 2-AG, 2-arachidonoylglycerol; CCl4, carbon tetrachloride; PLIN2, perilipin 2.
![]()
THE REGULATORY MECHANISMS UNDERLYING ROCK1-MEDIATED LIPOGENESIS
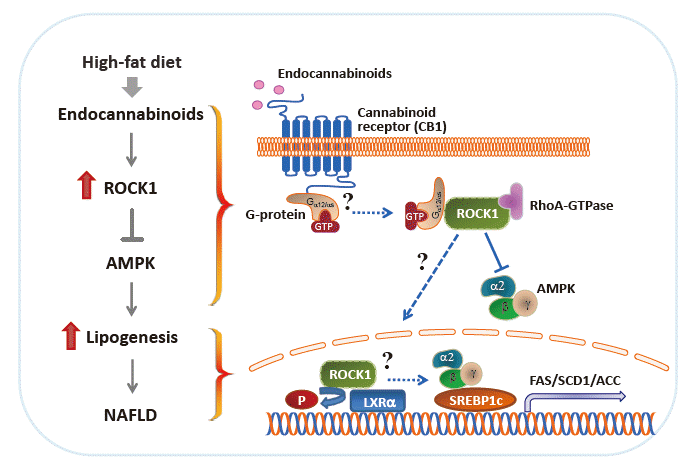
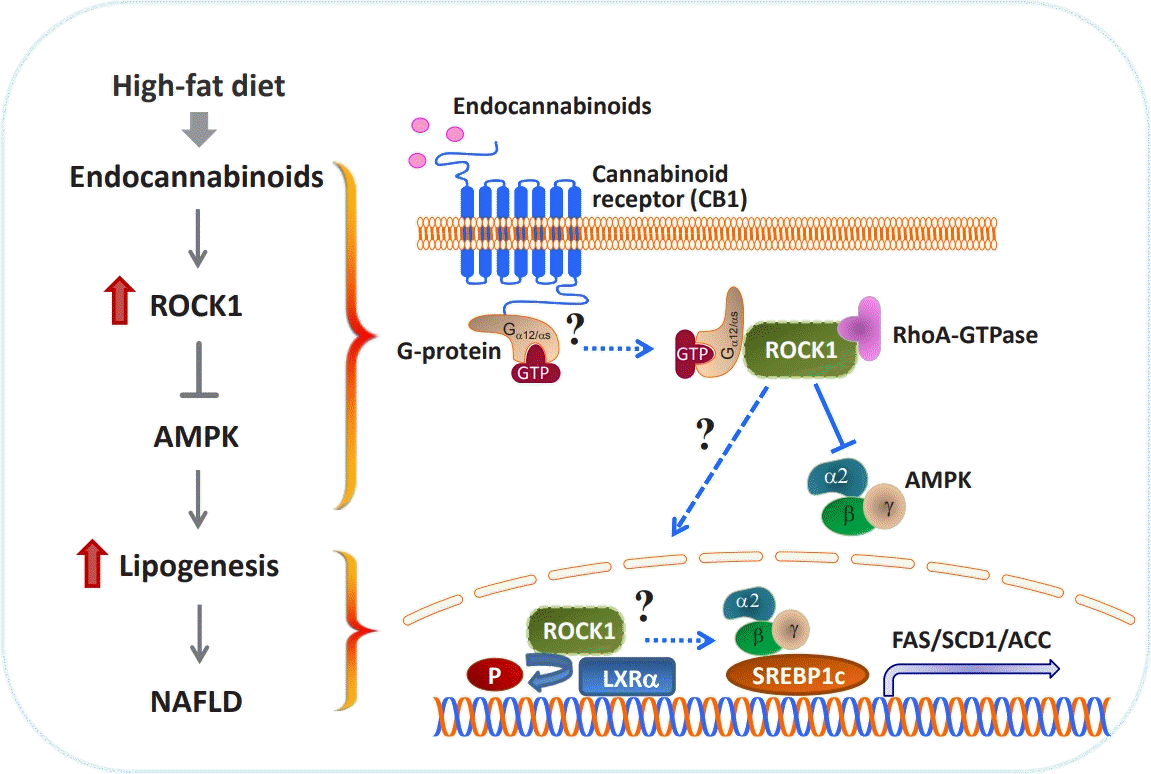 | Fig. 2.Role of Rho-associated coiled-coil-containing kinase (ROCK1) in the development of nonalcoholic fatty liver disease (NAFLD). High-fat feeding increases endocannabinoids 2-arachidonoylglycerol and anandamide levels produced from hepatic stellate cells of the liver. Endocannabinoids stimulate ROCK1 activity via cannabinoid receptor type 1 (CB1), which inhibits AMP-activated protein kinase (AMPK) activity. Suppressed AMPK activity then drives the sterol regulatory element-binding protein 1c (SREBP1c)-mediated lipogenic program, leading to a rise of lipid accumulation. NAFLD ultimately develops with the progression of obesity. On the other hand, it is hypothesized that activated ROCK1 binds to liver X receptor (LXRα) and may prompt the lipogenic pathway through SREBP1c. As an early event of ROCK1 signaling, CB1-mediated Gα12 (or Gαs) activation could be an initial step for ROCK1-dependent de novo lipogenesis. GTP, guanosine triphosphate; FAS, fatty acid synthase; SCD1, stearoyl-CoA desaturase-1; ACC, acetyl-CoA carboxylase.
|
 XML Download
XML Download