

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Diabetes mellitus increases the risk of developing diabetic cardiomyopathy, a specific cardiomyopathy first described by Rubler et al. [1]. Diabetic cardiomyopathy leads to heart failure in patients with diabetes that is independent of hypertension and coronary artery disease, and is characterized by diastolic dysfunction, ventricular hypertrophy, myocardial fibrosis, and cardiomyocyte apoptosis [1,2]. Although the mechanisms are not fully understood, overwhelming evidence indicates that cardiomyocyte apoptosis plays an important role in the development of diabetic cardiomyopathy [3,4].
Autophagy is a self-degradative process that removes protein aggregates and damaged organelles, and is important for balancing the sources of energy in development as well as in response to nutrient stress [5]. The majority of studies have indicated that autophagy is the most important regulatory target of cell survival, and it plays an important role in the development and prognosis of heart disease [6,7]. Zou and Xie [8] demonstrated that diabetes induces cardiomyocyte apoptosis and suppresses cardiac autophagy in diabetic mice. Moreover, recent studies have demonstrated, in experimental and clinical settings, that cardiomyocyte apoptosis is correlated with the inhibition of autophagy induced by hyperglycemia [9,10].
Granulocyte-colony stimulating factor (G-CSF) is a growth factor that mediates the proliferation, differentiation, and survival of hematopoietic progenitor cells as well as the mobilization of bone marrow cells [11]. Recent studies indicated that G-CSF improves cardiac function both after myocardial infarction and in dilated cardiomyopathy [12,13]. We previously reported that G-CSF treatment of diabetic rats improved cardiac diastolic dysfunction and attenuated cardiomyocyte apoptosis [14,15]. Therefore, in this study, we explored whether the mechanisms underlying the anti-apoptotic effects of G-CSF were associated with autophagy using a rat model of diabetic cardiomyopathy.
METHODS
Animals
Male Sprague-Dawley rats (Koatech, Pyeongtaek, Korea), aged 7 weeks and weighing 210 to 230 g, were used in this study. A combination of a high-fat diet (HFD, 60.3% of total calories come from fat, D12492; Research Diets Inc., New Brunswick, NJ, USA) and a low-dose of streptozotocin (STZ; Sigma-Aldrich, St. Louis, MO, USA) was effectively used to induce a rat model of diabetic cardiomyopathy [16]. The rats were maintained in a specific pathogen-free facility at the Hanyang University Medical School Animal Experiment Center under controlled conditions: temperature, 23°C±2°C; humidity, 55%±5%; and with an alternating 12-hour light/dark cycle. The experiments were performed in compliance with the animal research: reporting of in vivo experiments (ARRIVE) guidelines on animal research [17], and the research protocol was approved by the Hanyang University Institutional Animal Care and Use Committee (HY-IACUC-16-0107).
In vivo experimental design and drug treatment
The experimental design is outlined in Fig. 1. Seven-week-old rats were randomly assigned to one of two dietary regimens, either normal chow diet (n=6) or HFD (n=16) for an initial period of 7 weeks. After 6 weeks (at 13 weeks of age), the HFD group received a single intraperitoneal injection of STZ 30 mg/kg in 0.1 mmol/L citrate buffer, and the normal chow group received an injection of an equivalent volume of citrate buffer vehicle. One week later (at 14 weeks of age), fasting blood glucose (FBG) was measured, and rats with blood glucose level ≥200 mg/dL (11.1 mmol/L) were considered to have diabetes mellitus [18]. At 15 weeks of age, the diabetic group rats were randomly divided into two subgroups: diabetic rats treated with saline (n=8) and diabetic rats treated with G-CSF (n=8). Rats in the G-CSF treatment group were injected intraperitoneally with recombinant human G-CSF (200 μg/kg/day, Leucostim; Dong-A Pharmacological, Seoul, Korea) for 5 days. Rats in the normal chow group and the diabetic group treated with saline were injected intraperitoneally with an equivalent volume of saline for 5 days. At 18 weeks of age, all rats were euthanized for laboratory analysis.
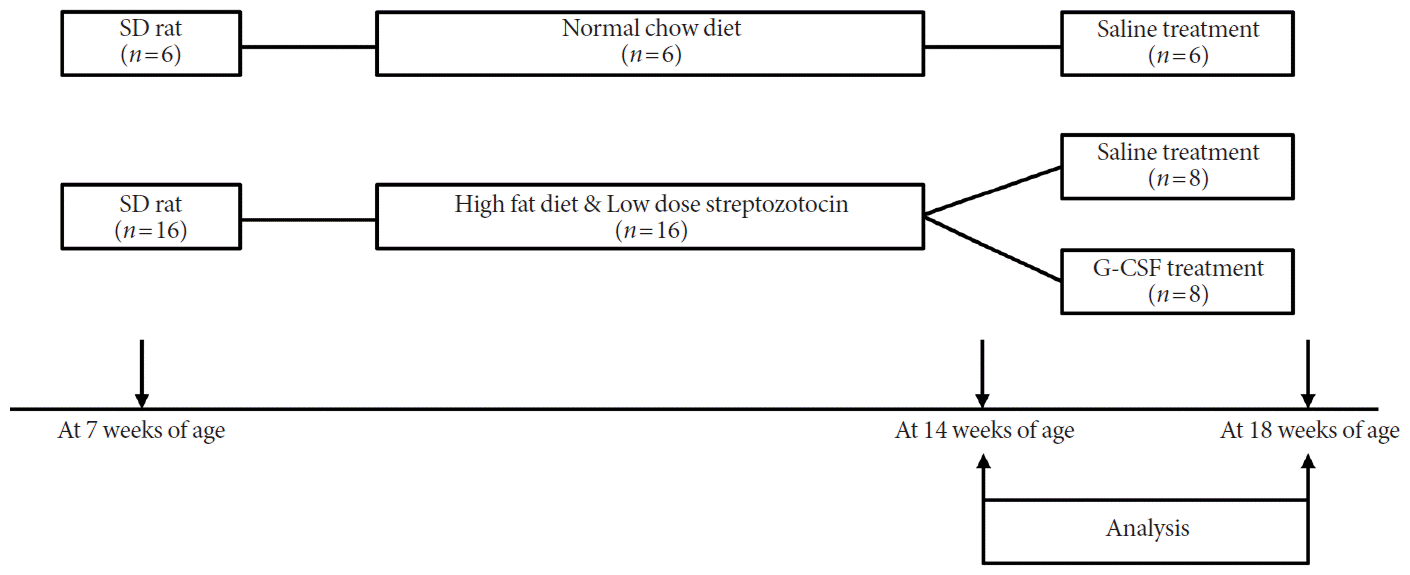
Fig. 1.
Scheme of the animal experiment. Diabetes was induced in rats by feeding for 7 weeks with a high-fat diet and low-dose streptozotocin (30 mg/kg) injection. Rat were then randomized for treatment with granulocyte-colony stimulating factor (G-CSF) or saline administrated intraperitoneally, for 5 days. Body weight, biochemical analysis, and echocardiography were performed both pre- and post-treatment. At 18 weeks of age, all rats were euthanized for histology and protein analysis. SD, Sprague-Dawley.
![]()
Body weight and biochemical analysis
Body weight, FBG, total cholesterol (TC), triglyceride (TG), and fasting insulin levels were measured. Blood samples were collected from tail veins after 8 hours of fasting. Serum glucose, TC, and TG were measured using an Olympus AU400 auto analyzer (Olympus GmbH, Hamburg, Germany). Fasting insulin was measured using an EZRMI 13K kit (Millipore, St. Charles, Mo, USA) according to the manufacturer’s instructions. Insulin resistance was estimated by the homeostasis model assessment of insulin resistance (HOMA-IR), using the following formula: HOMA-IR=fasting insulin (μU/mL)× fasting plasma glucose (mmol/L)/22.5 [19].
Echocardiography
Echocardiography was performed pre- and post-treatment. The rats were anesthetized by intramuscular injection of a mixture of zoletil 50 (30 mg/kg; Virbac SA, Carros, France) and rompun (10 mg/kg; Bayer Korea, Seoul, Korea) [20]. Serial echocardiographic examinations (VIVID E9; GE Healthcare with a 12 probe, Chicago, IL, USA) were performed by a single sonographer, with the rats in the left lateral decubitus position; the left side of the chest was shaved in order to obtain a clear image. The measurements included left ventricular ejection fraction (LVEF), early peak velocity of the early diastolic filling wave (E), and early mitral annulus velocity during the diastolic phase (E´) [14]. All measurements were based on the mean of five consecutive cardiac cycles; mean values were used in analyses.
Detection of myocardial apoptosis by TUNEL assay
Apoptotic cells in the myocardium were detected by the terminal deoxynucleotidyl transferase (TDT)–mediated dUTP–biotin nick end–labeling (TUNEL) assay in paraffin sections using an In situ Cell Death Detection kit (Roche, Mannheim, Germany). The stained sections were photographed using a light microscope (Leica DM 4000B; Leica Microsystems, Wetzlar, Germany). Five regions from each digitized image were selected at random, and the numbers of healthy and TUNEL-positive (apoptotic) nuclei were quantified. The apoptotic index was calculated as the number of TUNEL-positive nuclei/total number of nuclei [15]. All data were evaluated by an independent blinded investigator.
Cell culture
H9c2 cardiac cells (ATCC, Manassas, VA, USA) were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Life Technologies, New York, NY, USA) containing 5.5 mM glucose, 1% fetal bovine serum (Life Technologies), and 1% penicillin and streptomycin (Life Technologies) at 37°C in a humidified incubator containing 5% CO2 [21]. When cells reached 60% confluence they were divided into six treatment groups: (1) incubation with DMEM containing 5.5 mM glucose (normal); (2) incubation with DMEM containing 45 mM glucose (high glucose [HG]); (3) incubation with HG DMEM supplemented with 3 μg/mL G-CSF; (4) incubation with HG DMEM supplemented with 3 μg/mL G-CSF and 5 mM 3-methyladenine (3-MA; an autophagy inhibitor; Sigma-Aldrich, St. Louis, MO, USA); (5) incubation with HG DMEM supplemented with 5 mM 3-MA; (6) incubation with HG DMEM supplemented with 50 nM rapamycin (Sigma-Aldrich), an autophagy activator. After 36 hours, cells were harvested for flow cytometry and Western blot analysis of autophagy-related proteins. All tests were repeated at least three times.
Western blot analysis
The halves of the hearts were homogenized, and total protein was extracted using protein lysis buffer (Pro-prep; iNtRON, Seongnam, Korea). Cardiac tissue samples containing 60 μg total protein and H9c2 cell extracts containing 10 μg total proteins were boiled for 10 minutes and loaded onto sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels (8% stacking and 10%, 15% separating gels). Separated proteins were transferred to nitrocellulose membranes (NC, 0.45 μm pore size; Bio-Rad, Hercules, CA, USA) or Iimmobilon-P transfer membrane (PVDF, 0.45 μm pore size; Millipore, Billerica, MD, USA). After blocking in 5% bovine serum albumin solution (Sigma-Aldrich) or 5% skim milk solution (BD Biosciences, San Diego, CA, USA) for 60 minutes, the membranes were incubated with primary antibody overnight at 4°C. The primary antibodies used are specified in Supplementary Table 1. Blots were incubated with horseradish peroxidase-conjugated anti-rabbit antibody (1:2,000; Jackson Immunoresearch, West Grove, IA, USA) or anti-mouse antibody (1:2,000; Jackson Immunoresearch) for 1 hour at room temperature. Glyceraldehyde-3-phosphate dehydrogenase was used as a protein loading control. Positive protein bands were visualized using an ECL kit (GenDEPOT, Barker, NY, USA), and results were quantified with an image analyzer (Image lab 3.0; Bio-Rad).
Flow cytometry
H9c2 cardiac cells were cultured in 6-well plates and incubated with the appropriate drugs. Cells were resuspended in 500 μL of 1×binding buffer with 2 μL of Annexin V fluorescein isothiocyanate (FITC) and 2 μL of propidium iodide (FITC Annexin V Apoptosis Detection Kit I; BD Biosciences), and incubated at room temperature for 15 minutes in the dark. Cells stained with Annexin V-FITC and/or propidium iodide were analyzed by flow cytometry [22]. Three independent experiments were conducted for each condition investigated, with 1×104 cells analyzed per experiment.
Autophagic flux detection assay
Autophagy determination was performed using an Autophagy Detection kit (ab139484; Abcam, Cambridge, UK) according to the manufacturer’s protocol [23-25]. Autophagy Assay Kit ab139484 measures autophagic vacuoles and monitors autophagic flux in live cells using a dye that selectively labels autophagic vacuoles. The green dye accumulates in autophagy vacuoles based on the pH present in the vacuole. Moreover, it is pH clamed for pre-autophagosomes, autophagosomes, and autophagolysosomes. The quantity of stained vesicles reflects the degree of autophagy in the cell population. For flow cytometry, after treatment, cells underwent trypsinization and were pelleted. Cells were centrifuged at 125 rcf for 7 minutes to pellet the cells, then washed with 1x Assay buffer, and thereafter incubated with 250 μL of the diluted green stain solution for 30 minutes at 37°C in the dark. For fluorescence microscopy, cells were on 24-well plates on coverslips. After treatment, the medium was removed, and cells washed with 1x Assay buffer, following incubation with 100 μL microscopy dual detection reagent for 30 minutes at 37°C in the dark.
Statistical analyses
SPSS version 22.0 software (IBM Co., Armonk, NY, USA) was used for statistical analyses. All data are expressed as mean±standard deviation, except for histological and echocardiology data, which are expressed as mean±standard error. Data were analyzed using one-way analysis of variance (ANOVA) analysis (for multiple comparisons), and post hoc multiple comparisons were made with Tukey’s test (equal variances assumed) or Dunnett’s T3 test (equal variances not assumed). P values less than 0.05 were considered significant.
RESULTS
Body weight and biochemical analysis
At the end of the experiment, there were no significant differences in body weight between the diabetic and normal rats. The diabetic rats showed significantly higher FBG, TC, and TG levels compared with normal rats. The diabetic rats treated with saline also showed significantly higher HOMA-IR levels than normal rats, but there was no significant difference in the HOMA-IR level between the diabetic rats treated with G-CSF and normal rats (Supplementary Table 2). These results confirmed the successful development of a diabetic rat model using a combination of HFD and low-dose STZ.
Effect of G-CSF on cardiac diastolic dysfunction
Echocardiography was performed to assess cardiac function, pre- and post-treatment. At pre-treatment, LVEF was preserved, but the E’ velocity was significantly lower and the E/E’ ratio was significantly higher in diabetic rats than in normal rats, suggesting that the diabetic rats developed diastolic dysfunction. At post-treatment, echocardiography revealed that the E’ velocity was significantly higher and the E/E’ ratio was significantly lower in diabetic rats treated with G-CSF than in diabetic rats treated with saline, whereas LVEF and E velocity did not differ significantly between groups. In addition, there was a significant increase in E’ velocity (2.53±0.51 cm/sec vs. 4.00±0.55 cm/sec, P<0.05) and decrease in E/E’ ratio (27.35±5.01 vs. 17.19±2.13, P<0.05) in diabetic rats treated with G-CSF compared with pre-treatment (Supplementary Table 3). G-CSF significantly reduced the extent of fibrosis in the myocardium in diabetic rats, as observed in our previous study (Supplementary Fig. 1) [14,17]. Taken together, these results demonstrated that G-CSF has an ameliorative effect on diastolic dysfunction in a rat model of diabetic cardiomyopathy.
Effect of G-CSF on cardiomyocyte apoptosis in cardiac tissue
The apoptotic index was significantly lower in diabetic rats treated with G-CSF than in diabetic rats treated with saline (25.12%±4.24% vs. 34.51%±3.93%, P<0.05). However, there was no significant difference in the apoptotic index between diabetic rats treated with G-CSF and normal rats (Fig. 2A and B). To understand the molecular basis of increased apoptosis in the myocardium of diabetic rats, we measured the level of anti-apoptotic protein, B-cell lymphoma 2 (Bcl-2). The Bcl-2 protein level was significantly higher in diabetic rats treated with G-CSF than in diabetic rats treated with saline (82.86%±14.76% vs. 52.99%±19.58%, P<0.05) (Fig. 2C and D). These results suggested that G-CSF has an anti-apoptotic effect on the diabetic myocardium.
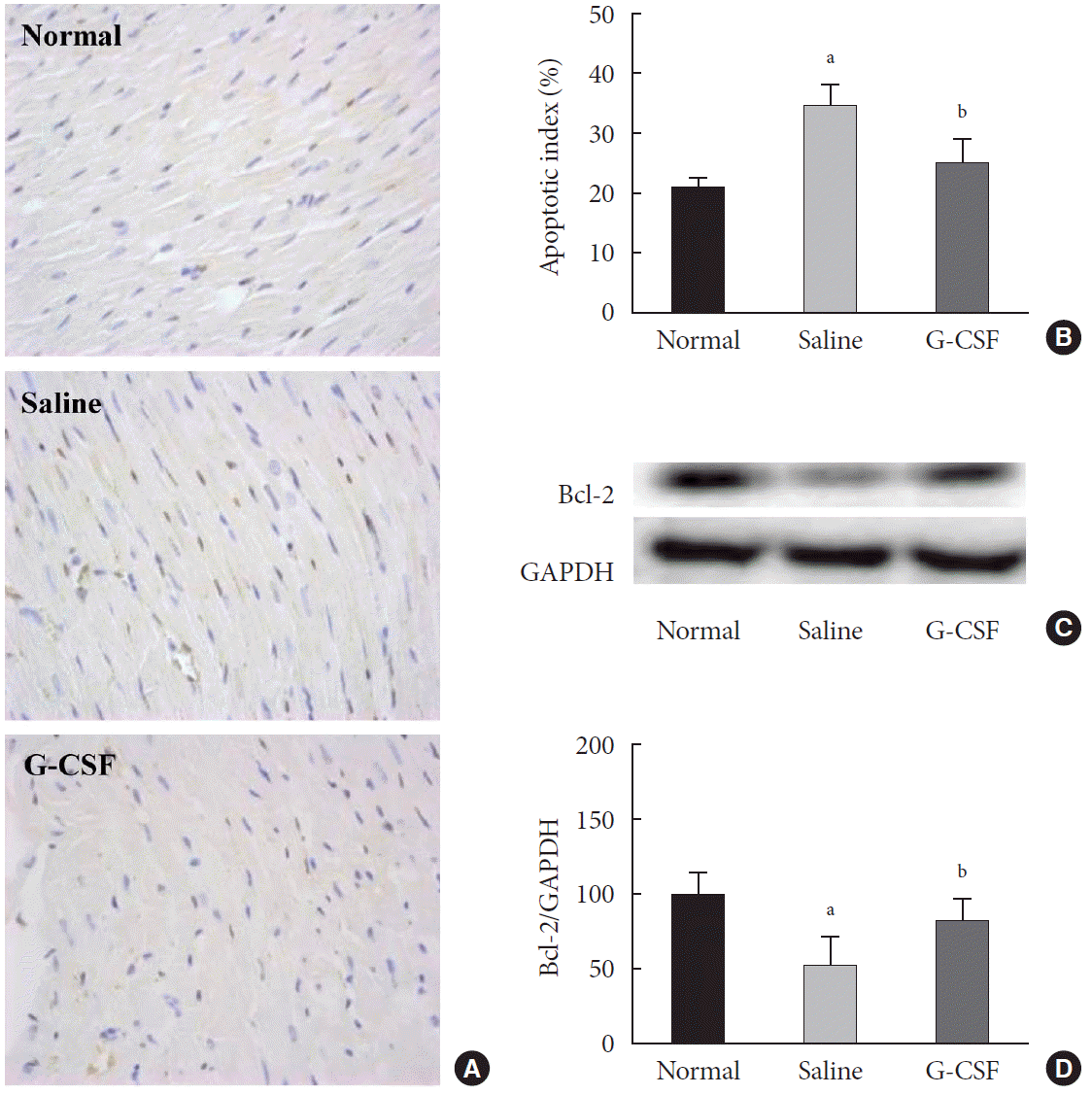
Fig. 2.
Effect of granulocyte-colony stimulating factor (G-CSF) on myocardial apoptosis in the diabetic myocardium. (A) Representative images of terminal deoxynucleotidyl transferase (TDT)–mediated dUTP–biotin nick end–labeling (TUNEL) assay staining of myocardium for each group 4 weeks after treatment (magnification ×400). Apoptotic nuclei are stained brown and non-apoptotic nuclei are stained blue on TUNEL assay staining. (B) Quantitative analysis of apoptotic cells in the myocardium of each group. (C) Level of B-cell lymphoma 2 (Bcl-2) protein in cardiac tissue was detected by Western blotting. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control. (D) Quantitative Western blot analysis of Bcl-2. The expression level was normalized by comparison with GAPDH expression. Protein levels are expressed as mean±standard deviation. Histology data are expressed as mean±standard error. aP<0.05 vs. normal group, bP<0.05 vs. saline group (n=6–8 per group).
![]()
Effect of G-CSF on autophagy in cardiac tissue
To clarify the effect of G-CSF on autophagy, we measured the cardiac Beclin-1 level, microtubule-binding protein light chain 3 (LC3)-II/LC3-I ratio, and P62 level, which are used as molecular markers of autophagy. Beclin-1 level was significantly higher in diabetic rats treated with G-CSF than in diabetic rats treated with saline (134.55%±25.46% vs. 70.08%±21.84%, P<0.05) (Fig. 3A and B). Moreover, the LC3-II/LC3-I ratio was also significantly higher (134.57±26.21 vs. 64.48±11.59, P<0.05) (Fig. 3A and C), whereas the P62 level was significantly lower (110.97%±13.85% vs. 169.56%±18.14%, P<0.05) (Fig. 3A and D), in diabetic rats treated with G-CSF than in diabetic rats treated with saline. These results suggest that the anti-apoptotic effect of G-CSF is significantly associated with upregulation of autophagy in the diabetic myocardium.
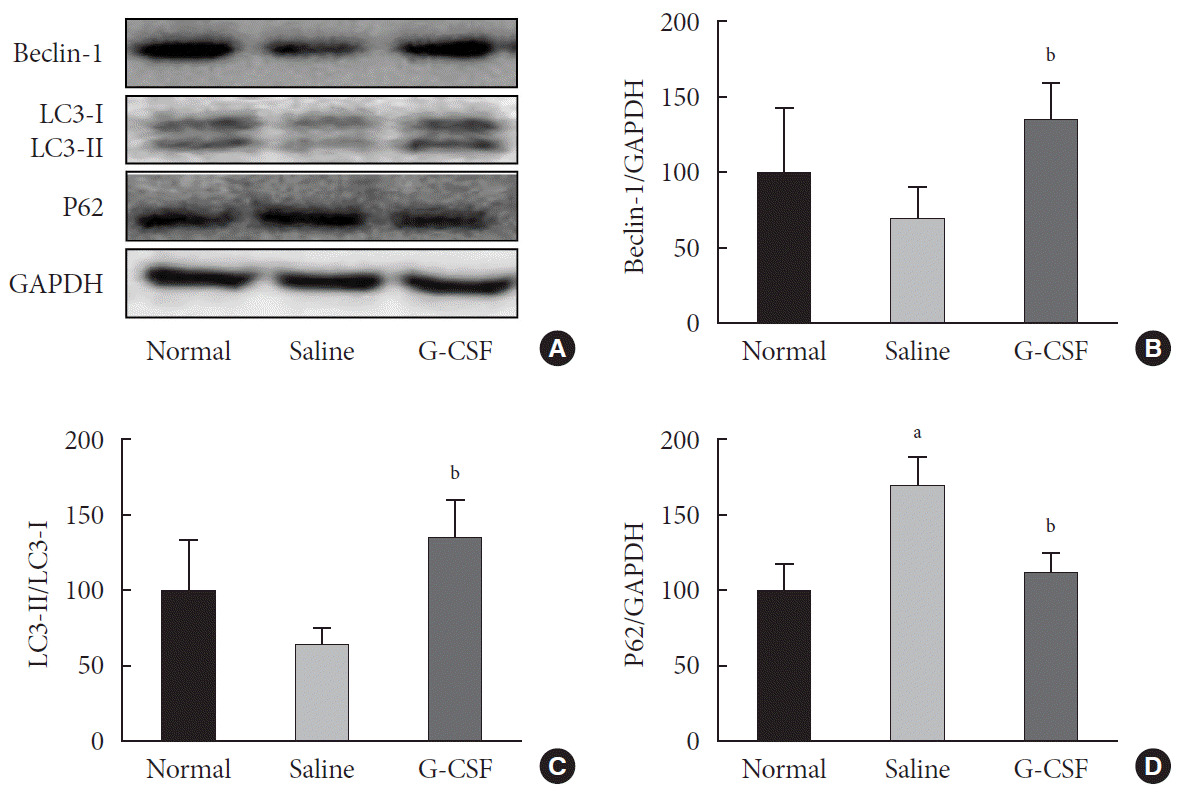
Fig. 3.
Effect of granulocyte-colony stimulating factor (G-CSF) on autophagy in the diabetic myocardium. (A) Representative images showing the levels of autophagy-related proteins Beclin-1, the microtubule-binding protein light chain 3 (LC3)-II/LC3-I ratio, and P62 in the diabetic myocardium measured by Western blot at 18 weeks of age (4 weeks after G-CSF and saline treatment). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control. (B, C, D) Quantitative Western blot analysis of Beclin-1, the LC3-II/LC3-I ratio, and P62. Protein levels were normalized by comparison with GAPDH expression. All data are expressed as mean±standard deviation. aP<0.05 vs. normal group, bP<0.05 vs. saline group (n=6–8 per group).
![]()
Effect of G-CSF on apoptosis in H9c2 cardiac cells
To clarify the effect of G-CSF on HG-induced apoptosis in H9c2 cardiac cells, we cultured H9c2 cardiac cells in HG media and measured the apoptosis rate by flow cytometry. HG media significantly increased the apoptosis rate of H9c2 cardiac cells compared with low glucose media (29.50%±3.90% vs. 16.50%±2.30%, P<0.05) (Fig. 4). Treatment with G-CSF significantly decreased the HG-induced apoptosis rate to 17.7% (Fig. 4). This effect was reversed to 33.1% by the autophagy inhibitor 3-MA (Fig. 4).
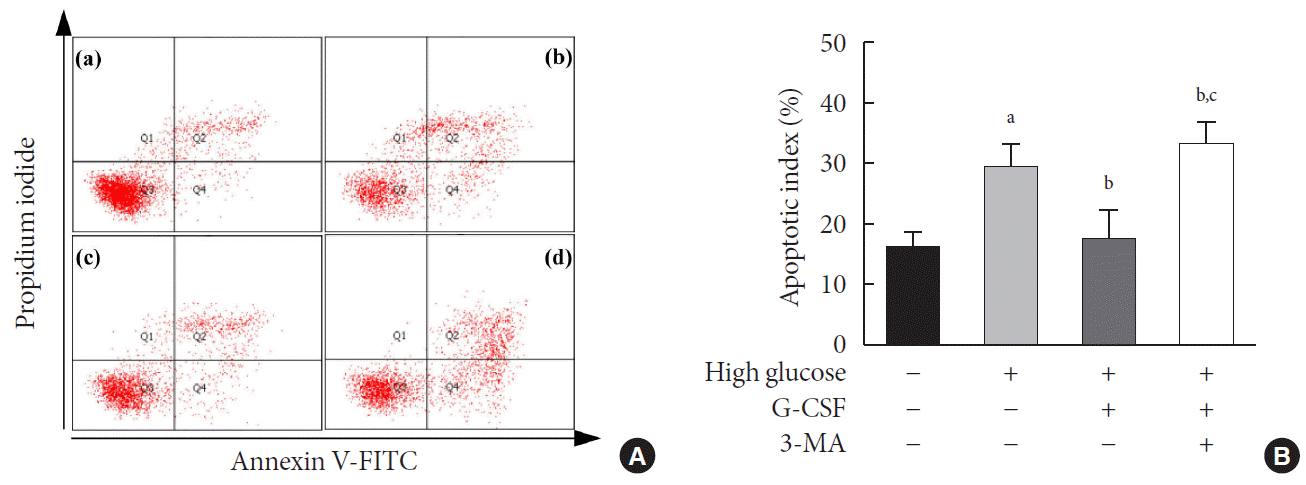
Fig. 4.
Effect of granulocyte-colony stimulating factor (G-CSF) on apoptosis in high glucose-induced H9c2 cardiac cells. (A) Dot plots displaying the stages of apoptotic death of H9c2 cardiac cells: Annexin−/PI− (Q3), viable cells; Annexin+/PI− (Q4), cells undergoing apoptosis; Annexin+/PI+ (Q2), cells in end-stage apoptosis or that are already dead; Annexin−/PI+ (Q1), cells that are in necrosis. (a) H9c2 cardiac cells cultured in low glucose media; (b) H9c2 cardiac cells cultured in high glucose media; (c) H9c2 cardiac cells cultured in high glucose media containing G-CSF (3 μg/mL); (d) H9c2 cardiac cells cultured in high glucose media containing G-CSF (3 μg/mL) and 3-methyladenine (3-MA; 5 mM). (B) Quantitative analysis of apoptotic cells (Q2+Q4). All data are expressed as mean±standard deviation. FITC, fluorescein isothiocyanate. aP<0.05 vs. H9c2 cardiac cells cultured in low glucose media, bP<0.05 vs. H9c2 cardiac cells cultured in high glucose media, cP<0.05 vs. H9c2 cardiac cells cultured in high glucose media containing G-CSF (n=5 per group).
![]()
Effect of G-CSF on up-regulation of autophagic flux in H9c2 cardiac cells
To investigate the effect of G-CSF on autophagy, we measured the Beclin-1 protein level, the LC3-II/LC3-I ratio, and P62 level. Treatment with G-CSF significantly increased Beclin-1 level (75.00%±5.37% vs. 50.41%±7.86%, P<0.05) and the LC3-II/LC3-I ratio (119.36%±14.37% vs. 75.07%±5.41%, P<0.05) in H9c2 cardiac cells cultured in HG media (Fig. 5A-C); these increases were reduced upon treatment with 3-MA (36.99%±3.06% vs. 75.00%±5.37%, P<0.05; and 51.01%±7.41% vs. 119.36%±14.37%, P<0.05) (Fig. 5A-C). The levels of P62 in H9c2 cardiac cells was significantly lower in HG media supplemented with G-CSF alone (68.57%±5.31% vs. 134.46%±19.55%, P<0.05) (Fig. 5A and D), but was increased by 3-MA (68.57%±5.31% vs. 144.35%±5.40%, P<0.05) (Fig. 5A and D).
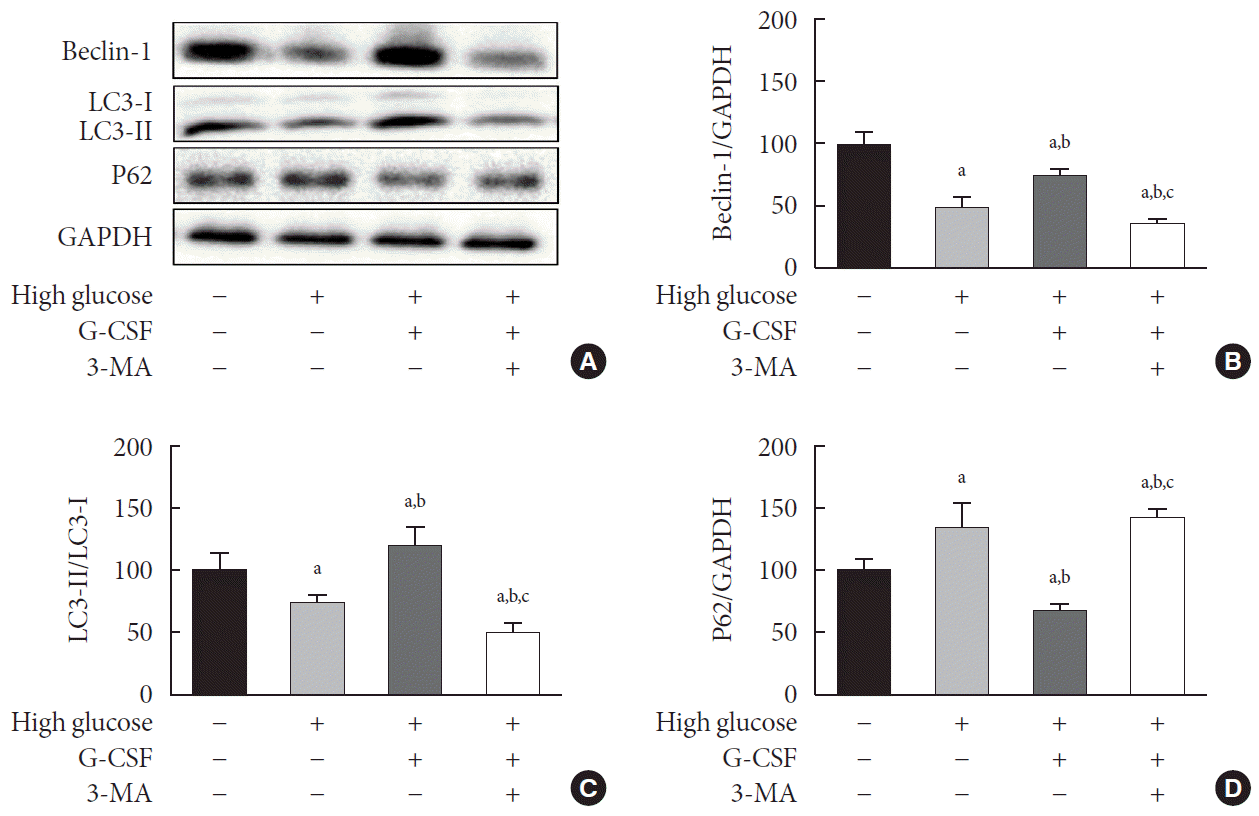
Fig. 5.
Effect of granulocyte-colony stimulating factor (G-CSF) on autophagy in high glucose-induced H9c2 cardiac cells. (A) Representative images of levels of autophagy-related proteins Beclin-1, the microtubule-binding protein light chain 3 (LC3)-II/LC3-I ratio, and P62 in H9c2 cardiac cells measured by Western blot. (B, C, D) Quantitative Western blot analysis of Beclin-1, the LC3-II/LC3-I ratio, and P62. Protein levels were normalized by comparison with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression. GAPDH was used as a loading control. All data are expressed as mean±standard deviation. 3-MA, 3-methyladenine. aP<0.05 vs. H9c2 cardiac cells cultured in low glucose media, bP<0.05 vs. H9c2 cardiac cells cultured in high glucose media, cP<0.05 vs. H9c2 cardiac cells cultured in high glucose media containing G-CSF (n=5 per group).
![]()
Furthermore, the apoptosis rate increased to 37.6% in H9c2 cardiac cells cultured in HG media supplemented with 3-MA, but decreased to 18.1% in cells cultured in HG media supplemented with rapamycin (Supplementary Fig. 2); this observation was consistent with the reduction in apoptosis in H9c2 cardiac cells induced by G-CSF under HG condition. In addition, the effect of G-CSF on autophagic flux in H9c2 cardiac cells was investigated using an Autophagy Detection kit. Using flow cytometry, we confirmed that treatment with G-CSF significantly increased autophagic flux (11.68% ±1.92% vs. 9.40%±0.45%, P<0.05) in H9c2 cardiac cells cultured in HG media (Fig. 6A and B). Additionally, using fluorescence microscopy, we showed that treatment with G-CSF increased the fluorescent signal, thus indicating enhanced autophagic flux in H9c2 cardiac cells cultured in HG media (Fig. 6C).
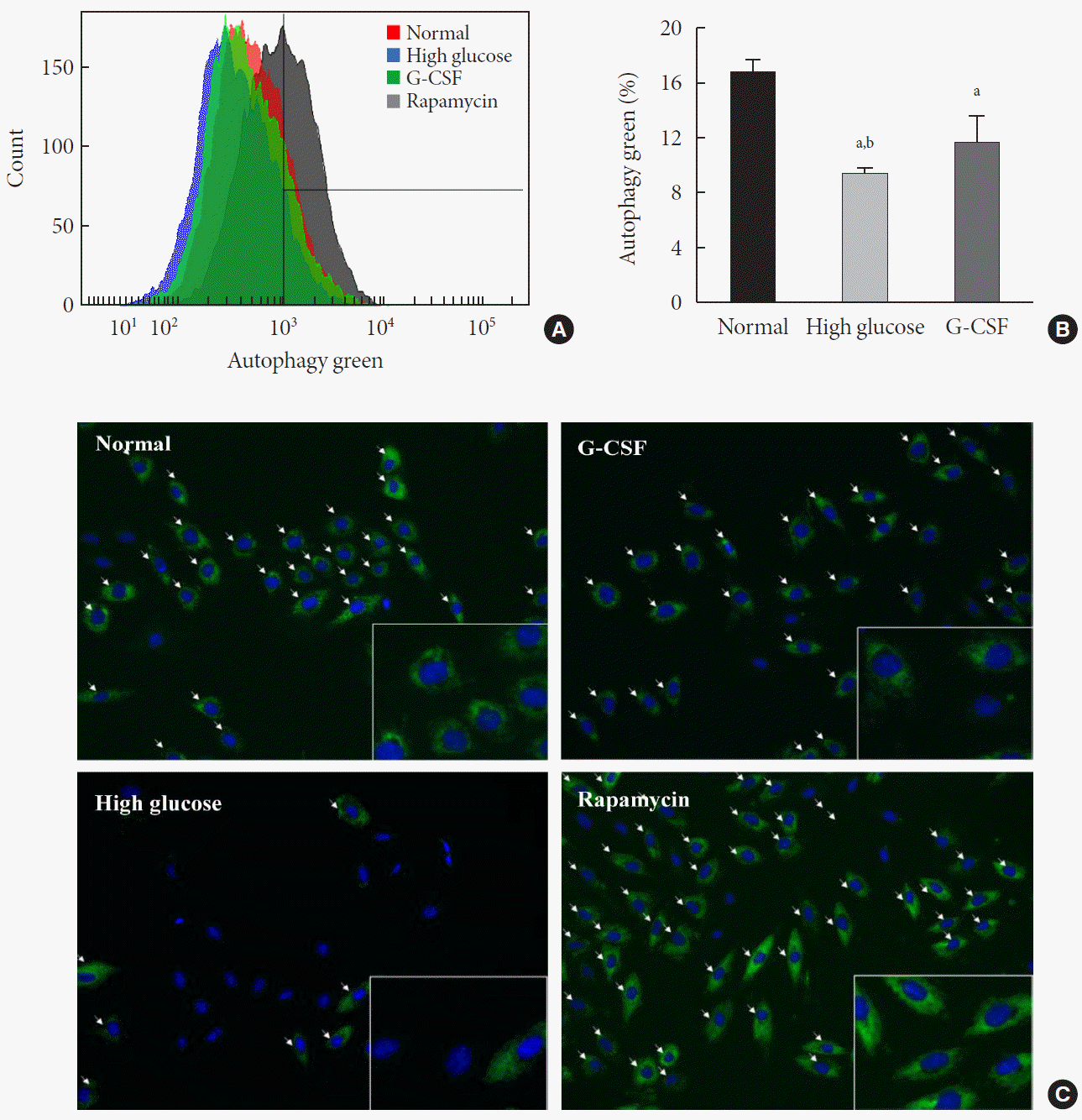
Fig. 6.
Effect of granulocyte-colony stimulating factor (G-CSF) on autophagic flux in high glucose-induced H9c2 cardiac cells. Autophagic flux evaluated high glucose-induced H9c2 cardiac cells by flow cytometry and fluorescence microscopy. Representative histogram (A) and bar graph (B), where fluorescence increase of autophagy green indicates autophagic flux increase. (C) Autophagic flux evaluated by fluorescence microscopy (magnification ×400), photos are representative of four to five independent experiments. Arrows indicate autophagic vesicles. The inserts in (C) show higher magnification. Rapamycin was used as a positive control of autophagy. All data are expressed as mean±standard deviation. Normal, normal condition group. aP<0.05 vs. H9c2 cardiac cells cultured in normal condition, bP<0.05 vs. H9c2 cardiac cells cultured in high glucose media containing G-CSF (n=4–5 per group).
![]()
Taken together, these results also suggest that the anti-apoptotic effect of G-CSF in H9c2 cardiac cells under diabetic conditions is linked to the up-regulation of autophagy.
DISCUSSION
The present study demonstrated that G-CSF reduced cardiomyocyte apoptosis and up-regulated autophagy in the diabetic myocardium. In addition, the anti-apoptotic effect of G-CSF in H9c2 cardiac cells under diabetic conditions was counteracted by treatment with an autophagy inhibitor. Moreover, we confirmed that G-CSF increased autophagic flux in vitro. These data indicated that the anti-apoptotic effects of G-CSF in a rat model of diabetic cardiomyopathy may be mediated by upregulation of autophagy.
Cardiomyocyte apoptosis is an important mechanism in the development of diabetic cardiomyopathy, which is closely associated with cardiac dysfunction, hypertrophy, and fibrosis [26,27]. In addition, several studies revealed that the reduction of cardiomyocyte apoptosis prevents diabetic cardiomyopathy both in animal models and in vitro experiments [28,29]. In previous experimental studies, G-CSF treatment reduced cardiomyocyte apoptosis by modulating apoptosis-related proteins [15]; however, the mechanisms underlying this antiapoptotic effect of G-CSF in diabetic cardiomyopathy remain unclear.
Autophagy is an important mechanism for cell survival, as it maintains the quality of proteins and organelles [30]. Studies have suggested that down-regulation of autophagy and the resulting accumulation of abnormal proteins and organelles, leads to apoptosis and cardiac dysfunction in various cardiac diseases, such as ischemic heart disease, cardiac hypertrophy, and heart failure [31,32]. Sishi et al. [33] and Wang et al. [34] demonstrated that up-regulation of autophagy prevented cardiomyocyte apoptosis in doxorubicin-induced cardiomyopathy and hypertensive heart disease, respectively. Moreover, overwhelming evidence indicates that cardiomyocyte apoptosis, which plays an important role in the development of diabetic cardiomyopathy, is induced by the impairment of autophagy [35-37].
The major finding in this study was that G-CSF increased Beclin-1level and the LC3-II/LC3-I ratio and decreased P62 level in the diabetic myocardium. Beclin-1 is an essential inductor of autophagy activity that binds to class III phosphatidylinositol 3-kinase to form a kinase complex in mammals [38]. LC3 is the major regulatory protein that promotes the induction of the autophagosome membrane [39]. When autophagy is initiated, LC3-I is conjugated to phosphatidylethanolamine to form LC3-II, which is required for the formation of the autophagosome [40]. P62 is another major factor that targets specific cargo for autophagy; P62 accumulates when autophagy is inhibited, and its levels decrease when autophagy is induced [41]. Consistent with our data, Zhao et al. [42] reported that heme oxygenase-1 up-regulated the expression of Beclin-1 and LC3-II in diabetic mice and suggesting that heme oxygenase-1 prevents diabetic cardiomyopathy by up-regulation autophagy. Moreover, activation of AMP-activated protein kinase was shown to protect cardiac structure and function by up-regulation of Beclin-1 and LC3-II expression, suggesting that increasing cardiac autophagy protect would protect cardiac structure and function in the diabetic myocardium [43]. In this study we showed that G-CSF up-regulated cardiac autophagy, as indicated by the increase in Beclin-1 level and LC3-II/LC3-I ratio and decrease in P62 level in the diabetic myocardium.
In this study, to confirm the effect of G-CSF on HG-induced apoptosis in cardiac cells, we cultured H9c2 cardiac cells with HG media, to create a diabetic cardiomyopathy model [44], and measured the rate of apoptosis using these cells. H9c2 cardiac cells are a commercially available myogenic cell line derived from embryonic rat ventricular tissue [45], which show cardiac-specific characteristics, such as morphological, biochemical, and electrophysiological characterization [46]. H9c2 cardiac cells offer a unique in vitro model to study the metabolic activity of the heart [47]. Moreover, we used 3-MA (an autophagy inhibitor) and rapamycin (an autophagy inducer) to further confirm that apoptosis of H9c2 cardiac cells was related to autophagy under diabetic conditions. We found that G-CSF reduced apoptosis of H9c2 cardiac cells, concurrent with the up-regulation of autophagy; these effects were abrogated by 3-MA. We also confirmed that inhibition of autophagy by 3-MA increased H9c2 cardiac cell apoptosis under diabetic conditions, whereas up-regulation of autophagy by rapamycin reduced H9c2 cardiac cell apoptosis, under diabetic conditions. Jia et al. [48] previously showed that safflower extract reduced apoptosis of H9c2 cardiac cells treated with angiotensin II, by increasing LC3-II expression. They also reported that the anti-apoptotic effect of safflower was reversed by 3-MA and that rapamycin reduced apoptosis, suggesting that safflower inhibits apoptosis via the up-regulation of autophagy. Gao et al. [49] similarly reported that 3-MA abrogated the anti-apoptotic effect of fasudil, suggesting that fasudil protects H9c2 cardiac cells from apoptosis via increasing Beclin-1 level and LC3-II/LC3-I ratio and decreasing P62 level under diabetic conditions. Guo et al. [50] also demonstrated that G-CSF promoted autophagy and reduced neural tissue damage after spinal cord injury in mice. Inhibition of autophagy by 3-MA partially blocked the neuroprotective effect induced by G-CSF, suggesting that G-CSF reduced neural tissue damage through up-regulation of autophagy. Moreover, we confirmed that G-CSF reduced the up-regulated autophagic flux under diabetic conditions. Considering our data and previous studies, we suggest that the anti-apoptotic effect of G-CSF is potentially mediated by the up-regulation of autophagy in H9c2 cardiac cells, under diabetic condition.
This study does, however, have several limitations. First, we demonstrated the effects of 3-MA and rapamycin in in vitro diabetic condition experiments, but we did not confirm the systemic effects of 3-MA and rapamycin in diabetic rats. In addition, we did not perform genetic knockdown or gain-of-autophagy methods that are necessary to determine the functional role of autophagy in the anti-apoptotic effects of G-CSF and to rule out the potential nonspecific effects of 3-MA and rapamycin that are unrelated to autophagy. Second, we were unable to investigate the down-stream signaling of autophagy-related proteins such as Beclin-1, LC3, and P62. Additional studies are required to further elucidate the detailed mechanisms regarding effect of G-CSF on apoptosis linked to up-regulation of autophagy. Third, we cannot rule out the possibility that the anti-apoptotic effect of G-CSF is associated with any other previously postulated mechanism such as the action of G-CSF directly or through the G-CSF receptor-mediated signaling pathway, a systemic effect, mobilization or homing of bone marrow stem cells, or other paracrine effects; such as fibrosis, vascularization, oxidative stress, or endoplasmic reticulum stress. Further studies regarding the precise mechanism of the anti-apoptotic effect of G-CSF are also worth exploring.
In conclusion, the results of our study indicate that the antiapoptotic effect of G-CSF may be significantly associated with the up-regulation of autophagy in diabetic cardiomyopathy. These findings suggest that G-CSF could potentially be used a novel therapeutic drug for the treatment of patients with diabetic cardiomyopathy.
 XML Download
XML Download