

 PDF
PDF Citation
Citation Print
Print



Introduction
Recent studies have shown that fetal status during pregnancy is related to future adult diseases. Exposure during pregnancy has been linked to lifelong health outcomes in children, including malnutrition and overnutrition, maternal mental illness, and substance abuse [1]. The major factors related to fetal programming are endocrinological disorders in pregnant women, infectious agents, toxins, and nutrient availability due to maternal nutritional status and placental functionality [2]. In the early stages of development, the body’s malnutrition “memory” becomes a pathology that determines future disease [3]. Fetal programming describes the fetus’s response to the intrauterine environment or the process by which it is affected by this environment. During this period of development, fetal programming can produce structural and functional changes in cells, tissues, and organ systems independently or through interactions with subsequent developmental processes and environments, with critical long-term consequences for future health and disease susceptibility [4]. In this review, fetal programming and the effects of fetal programming are discussed.
Go to : 
Fetal origin hypothesis
The ‘fetal origin of disease’ hypothesis proposes that hypertension, insulin resistance, dyslipidemia, cardiovascular diseases, and non-insulin-dependent diabetes in adulthood result from fetal adaptations to environmental malnutrition or placental insufficiency. These functional and structural changes in newborns likely develop at different times, particularly during pregnancy and very early childhood [5]. During the fetal period, body tissues and organs go through so-called “critical” developmental periods, known as rapid cell division. “Programming” describes the activity in which a stimulus or movement has permanent or lifelong effects during this critical developmental period [6]. Epidemiological studies have shown that there is a relationship between intrauterine environmental factors affecting fetal nutrition and the development of obesity and chronic diseases later in adult life [2]. The Developmental Origins of Health and Disease (DOHaD) hypothesis assumes that the prenatal period is a sensitive developmental period in which exposure to adverse environments as malnutrition, infection, or stress can have long-term or lasting effects on the health path of the offspring—this is termed “developmental programming” [7]. The “thrifty phenotype” hypothesis has been further developed to suggest that developmental programming is only one of a series of adaptations that occur in an external environment predicted during development. The “predictive adaptive response” theory, on the other hand, argues that predicting the environment in which the fetus will be born and preparing properly has selective advantages. The power of observation of these hypotheses is that diseases result from incompatibility between the fetal and postnatal environments [8].
Go to :
Fetal programming and epigenetics
Deoxyribonucleic acid (DNA) methylation plays a crucial role in embryonic and fetal development, particularly in establishing and maintaining cellular identity. In the first few days after fertilization, epigenetic marks from parent gametes are deleted to prepare them to receive cell- and tissue-specific marks, a dynamic phase of epigenetic reprogramming. This sequence of early developmental processes involving molecular mechanisms that maintain parentage of lineage-specific methylation in genomic imprints marks the peri-conceptional era for epigenetically mediated fetal programming effects [1]. Fetal programming during intrauterine life may be associated with maternal genes that influence fetal phenotype, independent of the fetal genome. Several studies in humans suggest that specific maternal genes may influence the fetal phenotype, regardless of the transfer of that particular gene to the phenotype. For example, maternal G protein β3 subunit (825T) [9], maternally expressed pleckstrin homology-like domain, family A, member 2 (PHLDA2) [10], CCNL1, and ADCY5 are strongly associated with birth weight via fetal genotypes [11]. These studies, with the general concept that genes of organisms that are originally from bacterial and viral infections can affect the physiology other organisms, support the advanced fetal programming hypothesis, which proposes that a maternal—and possibly paternal—gene in a human can affect the physiology of an offspring without being present in that particular individual [5].
Go to :
Maternal nutrition in the preconception period
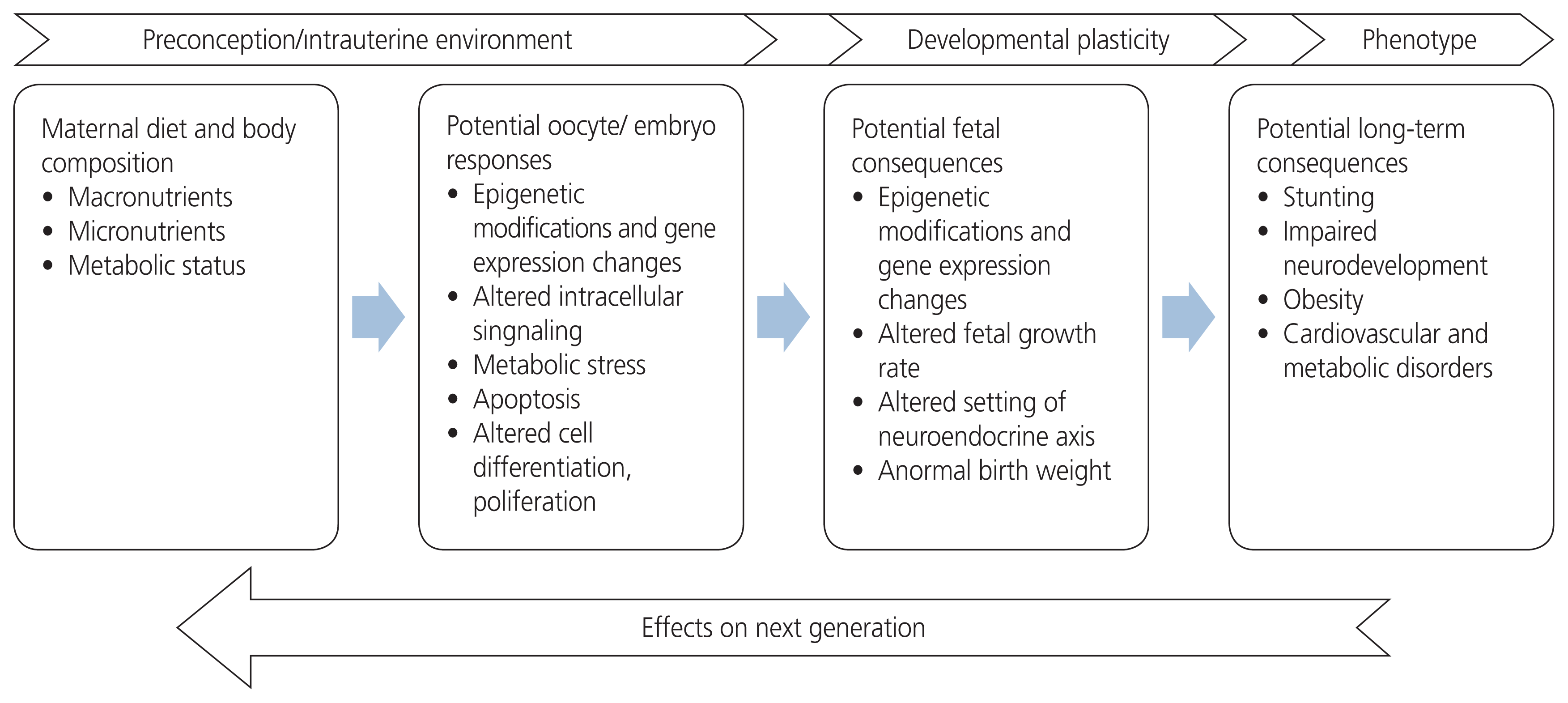
Eating well is essential for good health. The nutrition of expecting mothers is a fundamental public health problem because it influences not only the health of women but also the health of future generations. Health, lifestyle, and nutritional status in preconception periods are essential due to the consequences of gestational hypertension, preeclampsia, diabetes, and fetal growth restriction, which are important to monitor during pregnancy. These pregnancy outcomes affect offspring survival and increase the risk of developing non-communicable diseases in adulthood [12]. The origin of epigenetic modifications stems from maternal lifestyle choices, clues that exacerbate any pre-existing abnormalities in metabolism. Dietary quality and energy contents both affect human health in multiple ways and are inextricably linked to many metabolic states. The maternal diet contributes to a fetal “epigenetic signature” that affects the baby’s susceptibility to a risk of disease later in adult life [13]. The effects of nutrition on different generations are shown in Fig. 1.
Go to :
Maternal nutrition during pregnancy
Having a better nutritional status during pregnancy can meet the increased needs that result from pregnancy, and thus healthy pregnancy outcomes can be expected. In ideal nutritional conditions, foods should meet all needs, but fortified food and/or nutritional supplements may be recommended when a pregnant woman is thought to be malnourished [12]. Nutrition can affect DNA methylation through the action of the one-carbon pathway and its associated metabolites, including folate, riboflavin, choline, and betaine [1]. Maternal nutrition directly affects placental function and fetal development. Animal studies have shown that difficulties in the intrauterine environment activate the fetal hypothalamic-pituitary-adrenal (HPA) axis and disrupt fetal growth [14]. Results from cohort studies with humans suggest that an energy-dense diet, independent of maternal obesity, results in a hostile intrauterine metabolic environment that poses a risk. Therefore, future generations may encounter the consequences of energy-dense diets and obesity in mothers; thus, maternal diets and metabolism should be studied in order to understand the mechanisms that lead to an increased risk of type 2 diabetes [15].
The Pune Maternal Nutrition Study, in which 557 pregnant women participated, examined the different relationships between each parent’s height and body mass index (BMI) and fetal growth. Size and conditional growth results at 17 to 29 weeks were obtained from anthropometric measurements of the infant (ultrasound and head circumference, abdominal circumference, and femur length) and delivery measurements of placental volume (at 17 weeks only). Mothers with higher BMI values had fetuses with smaller average head circumferences at week 17; however, they later had greater head circumferences at birth. Placenta and fetal head growth are inversely related to maternal BMI at 17 weeks, possibly indicating that the prioritization of early placental development can be implemented as a plan to facilitate growth in late pregnancy [16]. Maternal obesity negatively affects brain development, behavioral status, and cognitive development of offspring. In animal studies, maternal obesity causes abnormalities in the hypothalamic and hippocampal areas and in the serotonergic, dopaminergic, and opioid systems, resulting in increased anxiety, impaired spatial learning and memory, and desensitization of the reward system [14]. Maternal malnutrition and protein-restricted diets have led to high blood pressure, insulin insensitivity, and renal dysfunction later in life. A high-fat diet in a mother leads to obesity and the development of diabetes in adult offspring. Studies on the impact of high fructose intake during pregnancy on the developmental programming of cardiovascular and metabolic dysfunction are still ongoing, with recent studies showing a link between a high-fructose diet and altered carbohydrate and lipid metabolism [17]. In a study with mice, it was found that mothers fed low protein diets exclusively during oocyte maturation resulted in changes in the behavioral phenotypes of their offspring and increased their susceptibility to cardiovascular disease. Similar findings have been reported in ewes, where maternal peri-conceptional energy restriction resulted in cardiovascular abnormalities, including increased heart rate and blood pressure [18].
Go to :
Intrauterine growth restriction
Intrauterine growth restriction (IUGR) means that the fetus does not realize its full potential growth. Placental insufficiency is a condition that can be managed and occurs due to factors such as maternal malnutrition, smoking, congenital infections, drugs, obesity, and chromosomal abnormalities [19]. The basic diagnostic criteria of IUGR include fetal weights below the third percentile for gestational age or low birth weights (LWs) below the tenth percentile (small for gestational age [SGA] and gender neutral) along with abnormal cord blood flow [20]. Early-onset IUGR is a critical situation characterized by disturbed uteroplacental and/or umbilical hemodynamics. Fetal distress could be end up with IUGR and is an indication for cesarean delivery [21]. Birth weight is determined by the intergenerational transmission of genetic material, the transmission of environmental factors, and the fetus’s responses to the uterine environment, which is also related to the birth weight of the previous generation due to fetal programming, which may affect the health of the fetus [22]. The fetus rearranges blood flow to nourish important tissues and organs, such as the brain, due to metabolic changes that slow down growth. It adapts by producing placental hormones such as fetal insulin and fetal insulin-like growth factor that control growth [23].
Go to :
The relationship between changes in fetal nutrition and adult diseases
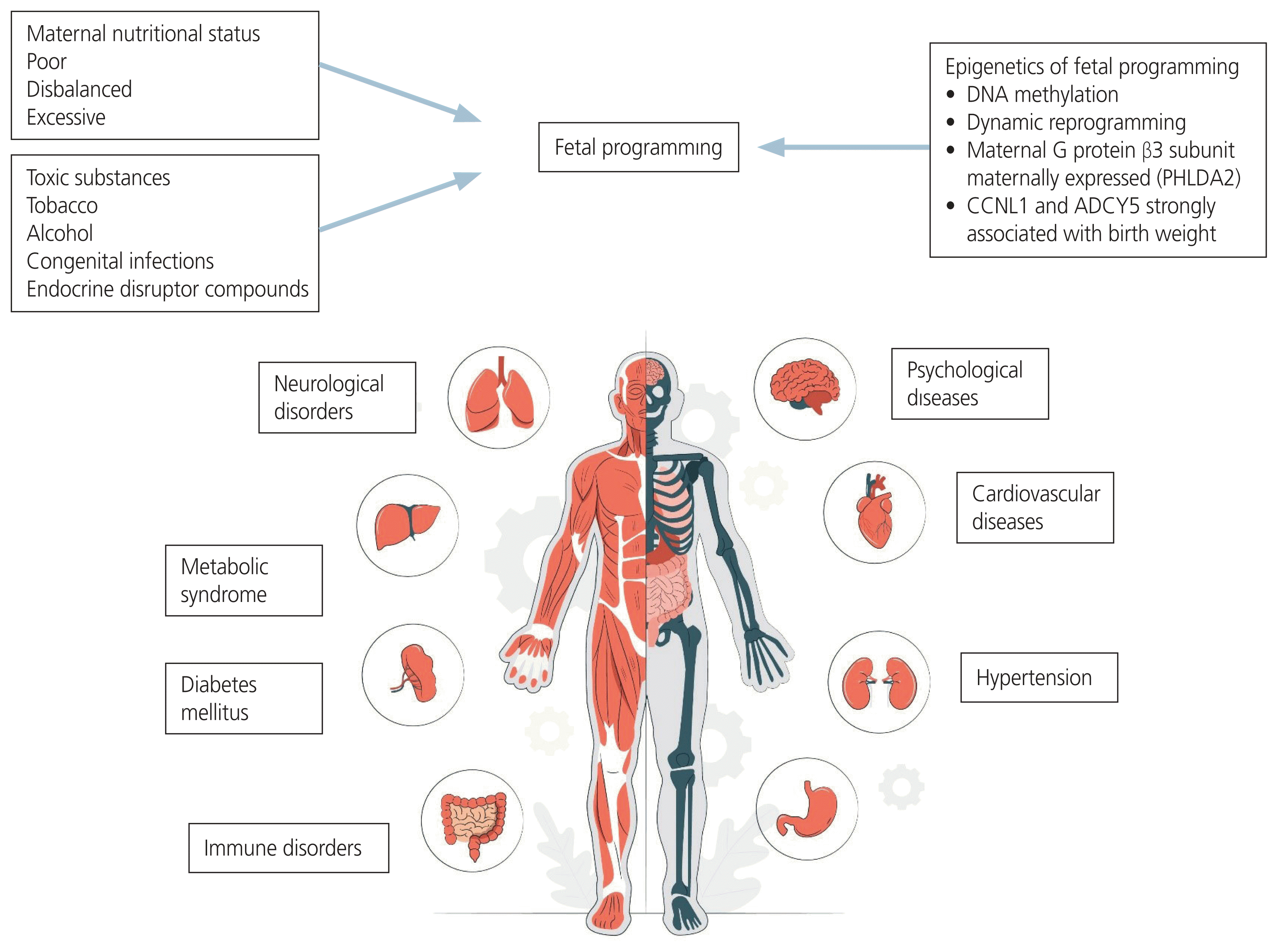
Observations from various cohorts have reinforced the DOHaD. In the historic Dutch famine that occurred after World War II, children born with IUGR due to famine had a higher prevalence of metabolic, cardiovascular, hepatic, renal, and neurocognitive impairments, as well as a higher incidence of type 2 diabetes mellitus (T2DM) [20]. Research has revealed the ‘fetal origin hypothesis’, which suggests that adaptations made by a malnourished fetus cause cardiovascular disease. These adaptations may result from the redistribution of metabolic, hormonal, or cardiac outputs in order to protect the brain from the most delicate organs, as well as from the slowing of growth in order to meet nutrient requirements. Compared to changes in human life patterns or treatments received during adulthood, certain situations to which a fetus is exposed during early development tend to have lasting effects on the structure and function of the body [24]. A summary of the effects of fetal programming on adult diseases is shown in Fig. 2.
Go to :
Cardiovascular diseases
A strong negative correlation between birth weight and the incidence of hypertension, non-fatal coronary disease, and stroke was found in extensive prospective cohort studies conducted in the late 1990s. Men were found to be more likely to have cardiovascular diseases than women born with IUGR. While this situation grew in men with childhood obesity in adolescence, it increased even more in those born to obese mothers. Rapidly gained postnatal catch-up growth, far from being beneficial, promotes the development of cardiovascular diseases and metabolic disorders in adulthood [20]. In the Helsinki Birth Cohort Study (HBCS), deaths from cardiovascular diseases (CVD) were associated with LWs. Men with a birth weight below 2,500 g had a hazard ratio for CVD of 3.63 compared to those with a birth weight above 4,000 g. To explain this situation, researchers hypothesized that people who are born small and experience slow growth in infancy have poor liver enlargement that predisposes them to dyslipidemia (a significant risk factor for coronary heart disease) later in life [25]. This unpredicted increase in risk was first observed in a Finnish cohort: mortality from CVD in adulthood increased in children with expeditious weight gain before reaching school age. The thrifty phenotype explains these observations [20]. The Atherosclerosis Risk in Young Adults (ARYA) study, which was conducted in 750 people born between 1970–1973, found that LW in the young adult period was associated with increased coronary artery disease [26]. It has been discussed that people whose growth is disrupted in their intrauterine and infancy environment may continue to be exposed to a hostile environment during their childhood and adult life and that the later environment will produce the effects attributed to programming [23].
Go to :
Diabetes mellitus
Gestational diabetes is known to cause adverse pregnancy outcomes. It is also strongly associated with long-term adverse effects on offspring that are the result of epigenetic modifications of the fetal genome [27]. To reduce oxidative stress and inflammation, which actively impair insulin sensitivity and pancreatic β-cell function in early pregnancy, the maternal metabolic environment needs to be normalized [28]. Insulin is a clear possible link due to its key roles in fetal growth, glucose and insulin metabolism disorders, premature growth, and cardiovascular disease. A sedentary lifestyle and obesity are thought to have a significant impact on the development of non-insulin-dependent diabetes. Although family and twin studies suggest familial predisposition, the nature of this phenomenon has not been fully explained. The disease is more likely to be transmitted by the mother than by the father of the family [23]. As stated by the “thrifty phenotype hypothesis,” decreased fetal growth causes a reduction in the number of pancreatic E cells and a decrease in the body’s ability to produce insulin, which is also associated with obesity that then leads to adulthood carbohydrate metabolism disturbances. The evidence that low-birth-weight newborns will have insulin resistance is strong. A systematic review published in 2008 by Whincup et al. [29] reported that birth weight was negatively correlated with the risk of T2DM and high blood pressure. A study of piglets found that LW offspring had significantly higher plasma levels of myoinositol and D-chiro-inositol, which matched glucose intolerance and insulin resistance results in adults [30]. In a randomized controlled study conducted on the children of mothers with gestational diabetes who were receiving diabetes treatment during pregnancy, it was found that normal-weight babies were born instead of macrosomic babies. However, the probability of being obese in these children after 5 years was the same as in macrosomic-born children [31]. Pregnancy-associated plasma protein-A (PAPP-A) releases insulin-like growth factor (IGF) and it is hypothesized that low PAPP-A levels in the maternal serum results with a reduction in active IGF levels. Inadequate serum IGF levels may affect the development of the embryo, and lead to other complications of pregnancy, such as stillbirth, risk of SGA neonates, preeclampsia, and premature delivery [32].
Go to :
Metabolic syndrome
Metabolic syndrome is characterized by several metabolic abnormalities, including type 2 diabetes and cardiovascular diseases. Metabolic syndrome is a combination of hypertension, hyperglycemia, high triglycerides, low high-density lipoprotein cholesterol, and central obesity. The fetal insulin hypothesis described the increased hazard of insulin resistance in adult life in babies born with IUGR. The fetal insulin hypothesis paradoxically suggests a contribution of genetic factors that alter fetal insulin secretion or the insulin sensitivity of organs. This is an example of the “thrifty phenotype” hypothesis: a suitable intrauterine environment can activate endocrine pathways and mechanisms that regulate gene expression, leading to decreased insulin secretion and increased insulin resistance [20]. It has been suggested that the developing fetus, vulnerable to an unfavorable intrauterine environment, may manifest invariant changes involving altered tissue physiology, hormone secretion, and glucose and lipid metabolism. When hyperglycemia is experienced during pregnancy, a proinflammatory reaction resulting in decreased blood flow and narrowing of blood vessels is triggered, leading to functional changes in placental cells. The same effect can occur with excessive glucocorticoid supplementation in response to intrauterine stress. Excessive amounts of glucocorticoids during the critical period of organogenesis cause a reduction in nephron counts, vascular dysfunction, persistent changes in hormone secretion, and angiotensin response inefficiency. These changes result in increased risk of hypertension and a predisposition to cardiovascular disease and hyperglycemia in later offspring development [2].
Hypertension
The relationship between LW and high blood pressure is more dependent on babies who are small for their gestational age after reduced fetal growth, as opposed to preterm babies [23]. The mechanisms that mediate the fetal programming of adult arterial hypertension are likely numerous. Several researchers have reported changes in kidney function due to a reduction in the number of nephrons, changes in the neuroendocrine system due to dysregulation of the HPA axis, and in the vascular system due to vascular activation and reduction of the density of arterioles and capillaries. To compare infants born at term through standard delivery methods and low-birth-weight infants, one study focused on the origin of vascular abnormalities and the endothelial angiogenic properties of colony-forming cells (ECFCs) present in the newborns’ umbilical cords. ECFCs are a type of cells defined by their ability to form colonies of endothelial cells and repair the physiological damage caused by changes in the vascular phenotype [33]. The role of the renin-angiotensin system (RAS) in the etiology of decreased nephron count and hypertension in fetal programming models was investigated. Components of the RAS are highly expressed in developing kidneys. The RAS plays a critical role in the nephrogenic period after birth in rats, as blockade of the angiotensin (Ang) type I receptor is associated with a decrease in renal function and an increase in arterial pressure, resulting in a reduction in nephron number [34]. In a study of 15,600 children aged 3 to 6 years, the risk of childhood hypertension was associated with greater birth a weight or postnatal weight gain, suggesting that intrauterine growth and postnatal weight gain may have implications for health during childhood [35]. A study conducted by Franco et al. [36] investigated the effect of birth weight and blood pressure on plasma levels of Ang-converting enzyme (ACE) activity and Ang and catecholamines in 66 children; they found that circulating noradrenaline was significantly higher in SGA girls than in appropriate-for-gestational age (AGA), Ang II, and ACE activity in SGA boys than in AGA boys.
Psychological diseases
Although the mechanisms by which maternal mental health affects fetal development remain unclear, its effect on the fetal and infant HPA axis develops through the programming of maternal cortisol. Additionally, the glucocorticoid system is also thought to play an important role in fetal programming. The development of the HPA axis and its associated capacity to regulate the stress response undergo significant changes during the fetal period and the first year of life, and its functionality is associated with many health outcomes. Accordingly, understanding the mechanism by which this happens is essential to understanding the programming pathways from mothers that affect the placenta, fetal development, and later child outcomes, including in terms of their effects on physical and mental health [37]. Short-term consequences of prenatal depression and anxiety on the physical health of pregnant women include obstetric complications and physical symptoms associated with lower fetal weight, limited neonatal growth, and decreased lower autonomic nervous system maturation. Studies have found that maternal prenatal depression and anxiety have similar effects on neonatal growth and behavioral and maturation outcomes. Newborns of parents with prenatal depression or anxiety had a higher risk of being born prematurely or having a LW, which are notable problems in infant health [38]. Lower birth weight for gestational age, placental infarction, neonatal hyporeactivity, and early eating difficulties were all significant predictors of bulimia development. Signs of delayed fetal growth (small and short for gestational age, short head circumference) and differences in fetal development measures suggest the impact of fetal programming on alternative metabolic, neural, and endocrine pathways in the development of anorexia nervosa versus bulimia [39]. The placenta, which can explain the etiology of some psychiatric diseases by providing morphogenic serotonin, is involved in metabolic pathways that modulate fetal brain development. Serotonin transporters (SERTs) play a role in autism spectrum disorder, and the mother’s SERT function alters offspring placental serotonin (5-HT) levels, suggesting that an altered placental 5-HT system can affect brain development [40]. In 2013, Freedman et al. [41] reported that LW plays a role in the neuropsychological deterioration seen in schizophrenia spectrum disorders in a study of low-birth-weight infants who reached the age of 40 years. He underlined that LW was associated with deficits in various domains, including working memory, general intelligence, and visuospatial deficits, and suggested that the youngest infants suffer from limited neural plasticity.
Go to :
Neurological disorders
According to epidemiological studies, exposure to excessive stress during the intrauterine period has the potential to adversely affect short- and long-term neurodevelopmental outcomes. While endocrine and immune stress mediators for neuronal and glial cell migration, differentiation, synaptic maturation, and brain development play a critical and essential role in many other pathways, unacceptable levels of these biological mediators can have detrimental effects on the developing brain [42]. Environmental signals can be transmitted from mother to fetus by influencing certain sensitive tissues and the physiological developmental course of the fetus during sensitive developmental stages, reshaping their structure and function, and thus reprogramming their susceptibility to diseases in postnatal life. They initiate a stigma process that permanently alters physiological systems, including the nervous system, predisposing them to premature aging and the onset of related diseases such as neurodegenerative diseases in the brain during adulthood [43]. Specific nutrient deficiencies or dietary imbalances can produce adverse neurodevelopmental effects, such as neural tube defects, language delays, decreased cognitive abilities, and mental and neurodevelopmental disorders. Studies on stress levels in pregnant women found that these women were higher risk for having offspring with neurodevelopmental disorders, affective disorders, and low cognitive abilities [4]. Fetal programming and stress can affect men and women differently. In humans, it has been shown that the placenta of female fetuses can provide relative protection from glucocorticoid excess due to increased glucocorticoid inactivation compared to men. Exposure to prenatal stress is associated with a higher risk of depressive symptoms, schizophrenia, and attention-deficit/ hyperactivity disorder (ADHD) in boys compared to in girls [44].
Go to :
Immune disorders
Once the mother’s immune system learns to accept the fetus during pregnancy, the developing fetus begins to form a separate immune system. Although placental and fetal membranes separate the mother and fetus, they are closely related physiologically and metabolically. While the precise mechanisms of the metabolic and physiological responses of IUGR are unknown, most non-communicable diseases result in immune dysregulation [45]. Metabolic diseases have also begun to be classified as inflammatory disorders due to the accompanying high concentrations of proinflammatory cytokines. Playing many important roles in the body, from regulating fetal growth and development, activating glucose and fat stores, controlling inflammation and modulating the immune response, glucocorticoids can alter metabolic function, leaving individuals susceptible to various adult metabolic disorders [46]. A meta-analysis investigating the association between birth weight and allergies found that IUGR protects against allergic diseases, which was consistent with preclinical evidence, although these results may differ between allergic diseases. Increases in birth weight were associated with higher odds ratios for food allergies in children and atopic dermatitis in children and infants, but not with allergic rhinitis in older children or adults [47]. Evidence from epidemiological and experimental studies suggests that IUGR reduces the risks of subsequent allergies, can lead to high methyl donor availability late in pregnancy or to active maternal asthma, and that allergies during pregnancy increase the allergy susceptibility of progeny [48].
Go to :
Endocrine disruptor compounds
The World Health Organization (WHO) has defined endocrine-disrupting compounds (EDCs) as exogenous substances or mixtures that alter the functions of the endocrine system and consequently cause adverse health effects in an intact organism, its progeny, or in (sub)populations.” Well-known EDCs are phthalate-based plasticizers or bisphenol A. Other sources of EDC include flame-retardants, pesticides, personal care products, and old compounds. They have deep chemical and biological stability and remain in the environment despite regulatory actions to eliminate or restrict their use and include arsenic, lead, chlorinated dioxins, dichlorodiphenyltrichloroethane, polycyclic aromatic hydrocarbons, polychlorinated biphenyls, and perfluoroalkyl compounds [49]. Given the rate of cellular proliferation, epigenetic programming, and many other developmental processes that characterize pregnancy, a healthy intrauterine environment is essential for optimal fetal growth. Exposure during this period of human life can adversely affect embryonic and fetal development [50]. The concept of fetal programming suggests that pre-pregnancy and/or prenatal and postnatal food and environmental chemical exposures have lasting and potentially adverse effects on future generations. Fetal nutrient requirements change over time, leading to dynamic metabolic adaptations in pregnant women [51]. According to the “environmental obesogen hypothesis,” exposure to obesogenic chemicals, particularly endocrine-disrupting chemicals (EDCs), including intrauterine life, may impair mechanisms related to adipogenesis or energy storage and therefore increase one’s susceptibility to being overweight. It interferes with growth trajectories early in life, as well as leads to obesity. Fetal growth retardation, fetal and maternal thyroid dysfunction, and postpartum neurological disorders may be associated with EDCs. EDCs are thought to interfere with the insulin, glucocorticoid, estrogenic, and thyroid hormone pathways and, therefore, may cause changes in the epigenome and inflammation state in later periods, with effects on endocrine and metabolic function as well as lifelong and even intergenerational consequences [52].
Go to :
Conclusion
Recent research has shown that all positive or negative experiences of the fetus during the intrauterine period can affect their future health status. Therefore, expectant mothers should be informed about fetal programming factors, and pregnancies should be planned accordingly. While planning a pregnancy, an expectant mother should undergo a detailed health screening; she should plan her nutrition, and if necessary, nutritional support should be provided. With education provided to expectant mothers, the health of the next generation could be preserved; this would lead to a decrease in the health system’s burden of chronic diseases and reduce their impact on the economy. Fetal programming should be considered to protect and improve adult health, and extensive research is needed in this area.
Go to :
 XML Download
XML Download