

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Hypertrophic cardiomyopathy (HCM) is characterized by hypertrophy of the ventricular wall and diastolic dysfunction with restrictive hemodynamic physiology. Histological findings of HCM include myocardial disarray with spatially disorganized fiber bundles with interstitial fibrosis. Calcium mishandling is typically reported to be the substrate for arrhythmia.
Pediatric HCM is distinguished from adult HCM in many aspects. Some children with HCM may be born with genetic mutations, and most of these patients do not present with onset of cardiac manifestations until the adolescent period. Although the onset ages of phenotypical presentations vary according to the individuals, the incidence of pediatric HCM peaks during the infantile and preadolescent periods. Among these, there is a group of infants with early-onset HCM who present with clinical signs in the first few months of life. These infants show massive progressive myocardial hypertrophy in only a few days or weeks with an astonishingly rapid speed, and they often have fatal clinical outcomes.1) This is not surprising to show that HCM diagnosed before 1 year of age generally accompanied to a worse outcome, with an approximately 30% rate of death or transplantation.2) The incidence of HCM is relatively low in young children and if the etiology of HCM is limited to idiopathic, the incidence is lower.3) Additionally, the incidence of infantile HCM may be underestimated because there is a high fatality rate during the infantile period and there can be sudden fatal cases of undetermined causes.
While ventricular wall hypertrophy in infantile HCM dramatically progresses in a short time, their symptoms and signs can be relatively vague at initial presentation with a non-specific history in their antenatal screening. Currently, pediatric HCM with gene mutations is often diagnosed, even before presenting symptoms, by genetic study, especially in cases with a family history of HCM among first-degree relatives.4)5) Similar to most major guidelines of screening for HCM from the adolescent period to adults, special surveillance for diagnosing infantile HCM is required.6)
Non-sarcomeric causes of HCM are more frequent in pediatric HCM, while sarcomeric causes are more common in adults. In addition, infants and children have more diverse causes of HCM than adults', including RASopathies, and pathological conditions such as metabolic, neurodegenerative, or mitochondrial diseases. Genetic causes such as mutations are also involved in. Mutations in the myosin-binding protein C3 (MYBPC3) gene are a frequent genetic cause of HCM7) and this typically shows onset of clinical presentation late in adolescence. Homozygous MYBPC3 mutant HCM is fatal during the early infantile period. Cases carrying a loss-of-function variant in a heterozygous state in either myosin heavy chain 7 (MYH7) or MYBPC3 with early-onset ventricular hypertrophy and left ventricular non-compaction, and deletion of the entire MYBPC3 gene show signs of their condition at 4 weeks after birth.8) A previous study showed that 20 Old Order Amish children with severe neonatal HCM caused by a homozygous mutation in the MYBPC3 gene presented at 3 weeks after birth and died during infancy.9)
To understand the distinct characteristics of infantile HCM, reviewing the maturation process of immature cardiomyocytes during the perinatal period is important. Recent decades have been a prominent era in developmental biology and understanding of cardiac development has increased. Cardiomyocyte turnover decreases exponentially with age and is <1% per year in adults. Total DNA synthesis as examined by genomic 14C incorporation in cardiomyocytes shows 2 peaks at <1 year of age and at approximately the prepubertal age.10) These time points are correlated with children's growth spurt with dynamic changes in hormones. The regenerative potential in partially resected newborn animals' myocardium has been investigated observing proliferation and cytokinesis of cardiomyocytes.11)
The mammalian fetal heart has immature cardiomyocytes, which are characterized by the naive property of proliferation during development, and they mostly exit their cell cycles shortly after birth, with limited turnover in adults.10)12)13) However, adult myocardium with heart failure reported to express fetal genes, and is often referred to as “fetal gene programming.” There is a much higher chance of regenerative capacity in neonatal cardiomyocytes compared with adult cardiomyocytes. Therefore, we can speculate on the characteristics of infantile HCM from the perspective of maturation of cardiomyocytes. Proliferation of cardiomyocytes extends into the first postnatal week in mice.14) In humans, the percentage of cardiomyocytes in mitosis and cytokinesis is highest in infants, and decreases to low levels by 20 years.15)
In clinical practice, phenotypes of HCM in neonates and infants are often encountered, but the pathophysiology is not clearly determined yet. Many observations could explain these phenomena. For example, perinatal period and early infantile period in premature infants correspond to dramatic hemodynamic changes with peripheral resistance. Metabolic changes are also dramatic and attributed to changes in oxygen, lactate, glucose, insulin, fatty acids, thyroxin, and glucocorticoids during the perinatal period. These factors have been comprehensively reviewed in articles underscoring the mitochondria as playing a central role in maturation of immature cardiomyocytes by producing sufficient ATP and sensing metabolic transitions, including oxygen and hormones.16)
Premature infants who are treated with steroids or neonates who are born to mothers with diabetes or hyperthyroidism often show phenotypes of HCM and these gradually resolve. These infants often show progression with an astonishingly rapid speed of hypertrophy in only a few days or weeks. The specific pathophysiology of these types of HCM phenotypes are not completely understood yet. Stressful environmental changes during the transitional circulation, various factors including fluctuations of hormones or hemodynamics delaying maturation of mitochondria along with premature birth, may affect maturation of cardiomyocytes.
With the recent remarkable advancement of molecular biology, understanding on the maturation of cardiomyocytes during development has greatly improved. This article reviews updated information on infantile HCM, including recent molecular biological and genetic studies on maturation of cardiomyocytes, from the clinical point of view to identify special characteristics of infantile HCM.
Go to : 
PROLIFERATIVE HYPERPLASTIC FETAL IMMATURE CARDIOMYOCYTES VERSUS NON-PROLIFERATIVE HYPERTROPHIC POSTNATAL MATURE CARDIOMYOCYTES
For improved understanding of infantile HCM, reviewing the updated basic concepts on maturation of cardiomyocytes is important. Cardiomyocytes undergo transition from fetal proliferative to postnatal hypertrophic properties with limited proliferative capacity after birth, which characterizes maturational hypertrophy. The regenerative capacity of cardiomyocytes gradually declines after birth. A murine study showed a rapid decline in ventricular cardiomyocyte cell cycle activity at birth followed by a burst of DNA synthesis in the first week of postnatal life by postnatal day 10 (P10).17) Additionally, there is a second burst during the preadolescent growth spurt when cardiac mass increases many folds over a few weeks and increases cardiomyocyte numbers by approximately 40%.18) In humans, the total percentage of DNA synthesis from 14C concentrations in cardiomyocytes shows that postnatal cardiomyocyte turnover is highest in the first decade of life and cardiomyocytes renew at a rate of 0.8% at the age of 20 years.10) However, in old subjects, this rate declines to values of <0.3%. While cardiomyocyte numbers are relatively constant throughout life after the adolescent period, endothelial and mesenchymal cells increase into adulthood and show a high turnover rate. Many reviews have updated hypertrophic growth of cardiomyocytes in the fetus to adults and cardiac regeneration based on proliferation and differentiation of cardiomyocytes.19)20) Proliferation of cardiomyocytes and progenitor differentiation contribute to heart growth at a young age. Cardiomyocytes that are positive for a mitosis marker increase in the first 20 years of life, after which activity is low.15) The important hallmarks of maturation of cardiomyocytes from immature fetal cardiomyocytes of a round and loose structure are characterized as follows:21) (1) myofibrillar maturation, including sarcomeric isoform switching of MYH from fetal to adult version; (2) maturation of electrophysiology and Ca2+ handling with expansion of the sarcoplasmic reticulum (SR) and T-tubule formation; (3) metabolic maturation with an increase in number and size of mitochondria; and (4) maturational hypertrophy by increasing the volume of cardiomyocytes with myofibrillar expansion and formation of intercalated disks. These maturation processes take time after birth and some of them require even a few years, especially regarding formation of T-tubules. In adults, when cardiomyocytes are injured, there are limited regeneration properties of these mature cardiomyocytes, and fibroblasts in the myocardium are activated to myofibroblasts in response to injury. This leads to scarring with pathological remodeling. However, immature cardiomyocytes act differently from mature cardiomyocytes. Murine and swine experimental models have shown complete repair by regeneration of cardiomyocytes after injury in the neonatal heart.11)14) In these models, regenerative capacity is lost by 7 days of age in mice and by 2 days of age in swine. After birth, the proliferation and regeneration capacity slows down and the turnover rate of cardiomyocyte at 20 years of age in humans is <1%.10) Therefore, this regenerative capacity after injury in animal models has focused attention on the window when cardiomyocytes proliferate during maturation by structural, transcriptional, and functional specialization. Sarcomeres are the key structure that regulate the maturation of the other organelles and the process of maturation of cardiomyocytes for the adaptation to pump efficiently after birth is complex.22)
With recent advancement of genetic tools, including CRISPR/Cas9 gene editing, cardiac studies have become comprehensive.23) Serum response factor (SRF), which is a key regulator of maturation of cardiomyocytes, was shown to be critical in perinatal maturation of cardiomyocytes.24) Additionally, this maturation is mediated in a stage-specific manner with absolute dependence on the balanced activity of SRF. In addition, a Cas9/AAV9-based somatic mutagenesis system in cardiomyocyte maturation was established.25) With this system, an alpha-actin-2 (Actn2) mutation results in defective structural mutation of T-tubules and mitochondria by triggering transcriptional dysregulation. Recent study identified an important role of Actn2-based sarcomeres in the regulation of maturation of cardiomyocytes with signal transduction and gene transcription.22) The molecular basis for HCM and congenital heart disease has been updated regarding genetic and epigenetic controls of heart development, highlighting specific maturation of cardiomyocytes.26)27) Factors affecting the transition from immature cardiomyocytes to hypertrophic mature cardiomyocytes have been described in many comprehensive reviews.20)21)27)28)
Go to :
PERINATAL HEMODYNAMIC CHANGES AFFECTING IMMATURE CARDIOMYOCYTES
Cyclic mechanical force that stretches cardiomyocytes induces differentiation and maturation of immature cardiomyocytes during development.29) Incessant stress by dynamic mechanical stress on immature cardiomyocytes contributes to maturation of neonatal cardiomyocytes by upregulating expression of cardiac markers, and affecting calcium handling and cell shapes. Activation of the cytoskeletal protein vinculin is essential for maturation of myofilament, which is related to this mechano-transduction pathway.30) The perinatal period is an essential transitional period for cardiomyocytes enhancing contractility to cope with the high workload of postnatal life. Although high oxygenation after birth causes maturation of cardiomyocytes, a hypoxic status in the fetal circulation delays myocardial maturation.31) Intrauterine growth restriction also delays maturation of cardiomyocytes in fetal sheep.32) This period may place a burden on the less organized immature myocardium with fewer sarcomeres per unit mass with different isoforms of contractile protein and fewer numbers of mitochondria. Extracardiac factors, such as a poorly compliant thoracic cage, changes in resistance of systemic and pulmonary circulations, closure of the ductus arteriosus, and a patent foramen ovale with increased arterial oxygen tension after birth place more burden on the left ventricle (LV) with preload and afterload. We often encounter cases of infantile HCM showing rapidly progressive hypertrophy of the LV in only a few days to weeks without any suspicious abnormality in antenatal screening images. Rapid progressive ventricular hypertrophy frequently occurs during changes in hemodynamics and peripheral resistance, which are aggravated by a hypoxic state due to lesions with immature lungs. The combination of multiple hemodynamic stresses may cause delayed transition from immature to mature cardiomyocytes and may be related to the pathophysiology of HCM in infants with unstable hemodynamics or those who are born prematurely.
Go to :
ELECTROPHYSIOLOGICAL MATURATION AND PROLIFERATION OF CARDIOMYOCYTES DURING THE PERINATAL PERIOD
HCM is characterized by enhanced cardiac contractility with impaired conformational changes during relaxation during the cardiac cycles. A previous study on adults with HCM investigated gene expression of calcium regulatory pathways using surgical myomectomy samples.33) This study showed that two major calcium modulators, sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) and calcium/calmodulin-dependent protein kinase type II, were dysregulated. In maturation of cardiomyocytes, electrophysiological maturation includes changes in ion channel type and localization with establishment of intercalated discs during the perinatal period. A study on murine ventricular cardiomyocytes showed that the SR progressively increases during postnatal development, whereas T-tubules are absent in the fetus and neonates, and only appear at 1 week after birth.34) A study on postnatal development of the energetic system controlling the ATP/ADP ratio in the murine heart showed that SERCA regulation progressively increased until 21 days after birth.35) With regard to human neonates, immature cardiomyocytes have underdeveloped SR and T-tubules. These immature cardiomyocytes rely on extracellular calcium concentrations necessary for muscle contraction in contrast to adult's cardiomyocytes, which have abundant intracellular calcium in the SR.
The myocardium shows adaptive hypertrophy in response to increased ventricular wall tension and neurohormonal stimuli by transiently increased vascular resistance during perinatal hemodynamic changes. Among the regulators of a hypertrophic response of the myocardium, calcineurin has been the focus of attention in studies of physiological cardiac hypertrophic signaling. HCM is associated with tacrolimus, which is a calcineurin inhibitor and acts as an immunosuppressive agent, as reported in infants and young children who are pediatric transplant recipients.36)37) Echocardiography has shown symmetric HCM with LV outflow obstruction, which resolves after stopping the tacrolimus. A previous study showed that altered myofilament Ca2+ sensitivity induced cardiac growth via generation of tension, inducing calcineurin and concentric remodeling, which could lead to cardiomyopathy.38) HCM mutations increase myofilament Ca2+ buffering, alter intracellular Ca2+ handling, and stimulate Ca2+-dependent signaling.39)
The effect of preterm birth on the developing myocardium in neonates with perinatal autopsy aged between 23 and 36 weeks of gestation was studied.40) Preterm birth resulted in a marked reduction in the proliferation of cardiomyocytes. This autopsy study of infants who died concluded that preterm birth appears to lead to an abrupt reduction in cell division of cardiomyocytes. However, a cardiac ultrasound imaging study showed disproportionate cardiac hypertrophy during early postnatal development in infants who were born preterm.41) Preterm infants at birth showed reduced cardiac mass relative to body size, but by 3 months, ventricular mass relative to body size was significantly higher than expected for postmenstrual age. A “critical perinatal window,” ranging from embryonic day 18.5 to postnatal 14 in mice, has been proposed during which the maturation process is exquisitely sensitive to perturbation.42) The review highlights SRF, peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) that potentially mediate complex dynamics of cardiomyocyte maturation. Further studies are required on the phenotype of HCM in the perinatal period, which is under electrophysiological and structural maturation.
Go to :
PHENOTYPE OF HYPERTROPHIC CARDIOMYOPATHY IN PREMATURE INFANTS WITH STEROID TREATMENT
In clinical practice, pediatricians often encounter cases of severe HCM in premature infants just after treatment with corticosteroids in the early days after birth (Figure 1). HCM is often diagnosed by chance with echocardiography while evaluating patent ductus arteriosus of premature infants, with rapid progressive ventricular hypertrophy in a few days, but this spontaneously resolves at a later time. Similar cases have been reported since the 1990s when postnatal steroid therapy was introduced as a standard therapy for premature infants.43) The Cochrane database on transient HCM in premature infants who are treated with corticosteroids for managing bronchopulmonary dysplasia (BPD) soon after birth (within 8 days) has shown that steroids increase the risk of neonatal hypertension and HCM.44)45)46) A recent meta-analysis on assessment of postnatal corticosteroids for preventing BPD in preterm infants with a gestational age of 32 weeks or younger showed that a moderately, early-initiated, high accumulative dose of systemic dexamethasone had a high risk of HCM in infants.47) The particular mechanism of transient LV wall hypertrophy in response to steroids in premature infants appears to be independent of dose or treatment duration.44)48)49)
 | Figure 1Echocardiographic images of a case of infantile HCM. Images were taken at 13 weeks after birth in a neonate who was born at the gestational age of 27+2 weeks to a mother with gestational DM. This neonate was treated with corticosteroids for severe BPD of a premature infant. Images were captured with a modified parasternal long-axis view owing to a limited transthoracic window with severe BPD. Massive diffuse LV wall hypertrophy (LV wall thickness z score: >10.0) in the systolic (A) and diastolic phase (B) can be seen.BPD = bronchopulmonary dysplasia; DM = diabetes mellitus; HCM = hypertrophic cardiomyopathy; LV = left ventricle.
|
Echocardiography has shown a distinct hypertrophic pattern of cardiac remodeling in preterm infants.50) Preterm infants have a significantly dilated, hypertrophied, and more spherical LV with impaired diastolic function compared with postmenstrual age-matched newborns at 36 weeks of gestation. However, the exact pathophysiology of this response to steroids of premature infants with transient myocardial hypertrophy has not been determined yet. For premature infants, steroids are usually prescribed in a recurrent hypoxic situation as with lung disease and a hemodynamically unstable condition, similar to a hypoxic fetal or perinatal transitional environment. This hypoxic environment may stimulate immature cardiomyocytes to proliferate in the ventricular myocardium. A rapid change in oxygenation change after birth is a major stimulus for cell cycle arrest of cardiomyocytes, resulting in conversion from hyperplastic to hypertrophic growth. Oxygen transition from fetal hypoxia to a postnatal oxygen-rich environment induces cell cycle arrest of cardiomyocytes as shown through a DNA damage response, while antioxidant treatment prolongs the postnatal proliferation capacity.51) A pathologically high level of oxidant species in cells has been suggested to drive progression of cardiovascular disease.52) Reactive oxygen species (ROS) are produced during oxidative phosphorylation and suppression of ROS may affect maturation of cardiomyocytes. An epitranscriptional mechanism of ROS is implicated in cardiac hypertrophy.53)
Dexamethasone-induced cardiac deterioration is associated with calcium handling abnormalities and activation of the calcineurin signaling pathway.54) Importantly, either insufficient or excessive glucocorticoid exposure before birth may alter the normal glucocorticoid-regulated trajectory of cardiac maturation. In the fetal and neonatal rat heart, antenatal dexamethasone leads to increased proliferation of cardiomyocyte, and glycogen synthase kinase-3β and β-catenin are thought to contribute to cardiac growth.55) Cardiomyocytes undergo cell cycle arrest when cardiomyocytes switch from the hyperplastic to hypertrophic mode of growth in the first postnatal week in rodents.56) However, according to the recent study, cardiomyocyte maturation events reported to occur over a 2-month postnatal period in swine model.57) In humans, 10% of postnatal day 1 cells are 2×2N and adult numbers of 2×2N cells are achieved within 1–2 weeks.15) Because preterm infants are born much earlier than the expected date, hyperplastic to hypertrophic transition of cardiomyocytes may not occur immediately after birth. Clinical cases of premature infants treated with corticosteroids who were complicated by the phenotype of HCM with LV outflow obstruction have been reported. This hypertrophy reaches a peak up to 3 weeks post-treatment and then resolves 1 month later,58) and often resolves completely several years later.59) These findings suggest that perinatal steroids affect proliferation of immature cardiomyocytes under maturation.
Go to :
HYPERTROPHIC CARDIOMYOPATHY IN INFANTS WITH MITOCHONDRIAL DISEASE
The onset and severity of cardiac presentation in mitochondrial disease remarkably vary depending on the patients. While some infants with mitochondrial disease die from fulminant heart disease at an early postnatal age, other children show gradual progression of cardiomyopathy at the adolescent period. Infants with mitochondrial disease present with non-specific clinical manifestations that are indistinguishable from other conditions, as sepsis. Mitochondria in immature cardiomyocytes also undergo a maturational process during myocardial maturation morphologically and functionally to increase production of ATP for coping with a highly demanding postnatal hemodynamic burden. However, mitochondria are regarded as central mediators triggering maturation of cardiomyocytes during the metabolic transition in cardiomyocytes.16) These findings suggest that normoxia, enteral feeding, and hormonal changes during the perinatal window affect mitochondria, and mitochondria can regulate cell cycle activity and electrophysiological properties of cardiomyocytes.
The mitochondrial system in skeletal muscle of preterm and full-term neonates with a gestational age of 25–42 weeks was studied.60) This study showed that mitochondrial parameters were lower in neonates who died within the first week of life than those aged older than 5 years. Additionally, enzyme activity was significantly lower in very preterm infants at an age of 1–7 days than those aged from 3–8 weeks, which suggested postnatal maturation of mitochondrial energy metabolism. Similar to the heart in which the energy substrate switches from glucose to fatty acids after birth, a role for PGC-1α on transcriptional control of mitochondria during postnatal cardiac growth has been implicated in a developmental stage specific. Germline disruption of PGC-1α and PGC-1β genes results in perinatal arrest of mitochondrial content at day 1 after birth and lethal early with cardiomyopathy relating to the perinatal maturation of the heart.61)
Mitochondria have been the focus attention in maturation of cardiomyocytes and mitochondrial biogenesis and dynamics in the developing and hypertrophied heart.62) A recent review integrated changes in cardiac metabolism and pathological remodeling with changes in cell phenotypes.63) Fetal cardiomyocytes rely on glycolysis for energy with low mitochondrial abundance. Cardiomyocytes need to be mature to switch from glycolysis to oxidative phosphorylation to cope with the postnatal environment. Inhibition of the switch to oxidative metabolism in the neonatal period promotes pathological remodeling. Gene mutation of acyl-COA dehydrogenase 9 (ACAD9), which is a mitochondrial protein involved in oxidative phosphorylation complex 1 deficiency, also shows fulminant infantile HCM (Figure 2). Patients with ACAD9 mutation die in the infantile or childhood period.64)65) Cases of ventricular non-compaction cardiomyopathy may not present until later in life, and fulminant neonatal presentation of HCM with non-compaction in Barth syndrome are often reported. A previous study on patient-derived and genetically engineered induced pluripotent stem cells determined the pathophysiology of Barth syndrome.66) This study highlighted the mitochondrial role in cardiomyopathy and showed that excess levels of ROS mechanistically linked TAZ mutation with impaired function of cardiomyocytes. This study showed the exciting results that the cardiomyopathic phenotype in Barth syndrome was readily reversed by reintroduction of wild-type TAZ or by suppression of excessive ROS produced by Barth syndrome mitochondria.
 | Figure 2Echocardiographic images of an 8-month-old infant with ACAD9 gene mutation. The patient had a birth weight of 2.9 kg at 37 weeks of gestation and showed developmental delay. Antenatal sonography showed non-specific findings. These images show a diffuse hypertrophied left ventricular wall on the parasternal long-axis view (A) and apical four-chamber view (B) with a thick interventricular septum (z score: 10.0).
ACAD9 = acyl-COA dehydrogenase 9.
|
Go to :
PHENOTYPE OF HYPERTROPHIC CARDIOMYOPATHY IN INFANTS BORN TO DIABETIC MOTHERS
The phenotype of HCM in neonates is frequently observed in neonates born to mothers with diabetes mellitus (DM). Cardiac hypertrophy is reported to affect 50% of infants of mothers with type 1 DM and 25% of those of mothers with type 2 DM.67) The exact mechanism of this finding has not been precisely defined, but a compensatory increase in fetal insulin secretion with diabetic mothers is a mechanism of HCM in neonates.68) Hyperinsulinism is widely associated with cardiac hypertrophy, which is indistinguishable in echocardiography from other pathological types of HCM.69) Infants of mothers with DM also have an increased risk of persistent pulmonary hypertension of the newborn, which is aggravated by hypoglycemia, asphyxia, and respiratory distress syndrome. The fetal heart primarily depends on glucose and lactate for energy sources in the hypoxic intrauterine environment because circulating fatty acids are low owing to poor placental transfer during the fetal period.70) Glucose uptake in the fetal heart depends on insulin. Infants born to mothers with diabetes develop postnatal hypoglycemia, macrosomia, and HCM in severe cases. Insulin-like growth factors 1 (IGF-1) and 2 are regulators of fetal and postnatal growth, and there have been studies on insulin signaling pathways and cardiac growth.71) Hyperinsulinism resulting from an absence of insulin receptor, acting through the IGF-1 receptor, causes HCM along with intrauterine growth restriction in Donahue syndrome.72) Increased IGF-1 levels at the time of delivery are correlated with neonatal septal HCM in infants of mothers with diabetes.73)74) Reduced glucose uptake in the postnatal heart indicates that facilitated maturation of cardiomyocytes and high levels of glucose may inhibit maturation of cardiomyocytes at genetic, structural, metabolic, electrophysiological, and biomechanical levels by promoting excessive nucleotide biosynthesis through pentose phosphate phosphorylation.75) High glucose levels suppress and delay cardiac maturation, promoting excessive proliferation with increased mitotic activity.75) In the experimental gestational DM model, fetal exposure to maternal hyperglycemia by infusing glucose to the left uterine artery late in gestation induced septal hypertrophy and myocardial proliferation compared with un-infused control littermates.76) With regard to the high reliance of the developing heart on glucose for energy, changes in glucose metabolism on pathological cardiac remodeling and their clinical effect have been discussed in comprehensive reviews.63)77) The phenotype of HCM in a neonate from a mother with DM might temporarily present with proliferative immature cardiomyocytes, which is expected to gradually resolve with maturation of cardiomyocytes when infants grow older.
Go to :
PHENOTYPE OF HYPERTROPHIC CARDIOMYOPATHY IN INFANTS BORN TO MOTHERS WITH HYPERTHYROIDISM
HCM in neonates born to mothers with hyperthyroidism often do not have long-term complications. Maternal thyroid function affects growth and maturation of fetal organs.78) Maternal hyperthyroidism is associated with neonatal thyroid disease, intrauterine growth restriction, low birth weight, and cardiomyopathy, with a prevalence of <1%.79) IGF-1 stimulates proliferation of fetal cardiomyocytes, while thyroid hormone inhibits proliferation and drives maturation of the cells.
Fetal circulating tri-iodo-thyronine (T3) levels are related to maturation of fetal organs. A previous study showed that T3 levels surged near term and T3 directly affected maturation of cardiomyocytes.80) This study included the potential effect of thyroid hormone on immature cardiomyocytes during the perinatal period with data from a fetal sheep model, which shares a similar growth trajectory of the human heart. In fetal sheep, increased circulating T3 levels promote terminal differentiation, with an increased proportion of binucleated and hypertrophied LV cardiomyocytes.81) Serum T3 levels abruptly increase at birth.82) Either high or low T3 levels alter the growth patterns of cardiomyocytes, but to what degree they affect maturation is unknown.80) Developmental trends in cord and postpartum serum thyroid hormones in preterm infants have also been reported.83) Postnatal free T4 levels are lower in premature infants and those aged 23–27 weeks of gestation are hypothyoxinemic in T4 levels compared with term infants. Similar to a direct effect of T3 on development of cardiomyocytes, a pathological hormonal change of the thyroid delaying normal transition to postnatal maturation may lead to transient HCM during the infantile period. Pathological hypertrophy in the neonatal heart is related to complex hormonal changes during the transitional circulation, and further related studies of this issue are required.
Go to :
HYPERTROPHIC CARDIOMYOPATHY IN INFANTS WITH NOONAN SYNDROME
Noonan syndrome (NS) is characterized by distinctive craniofacial features and is often encountered in clinical practice with HCM in neonates or infants. The RASopathies are caused by pathogenic genes encoding proteins related to the RAS/mitogen-activated protein kinase (MAPK) pathway. RASopathies overlap many phenotypic features.84) NS has cardiovascular abnormalities in 80–90% of cases and HCM is the second most commonly associated lesion.85) The most common gene associated with NS is PTPN11, which accounts for approximately 50% of all cases.86) HCM in infants with NS requires special attention because it is fulminant and progressive during infancy. Children with NS who are diagnosed with HCM before 6 months old more frequently (51%) present with heart failure than other children with HCM, and have a worse risk profile, resulting in early mortality (22% at 1 year).87) Decreased height for age and a lower LV fractional shortening z score on echocardiography are independent predictors of mortality in NS with HCM.87) Therefore, introduction of aggressive therapy for this condition, including early cardiac transplantation, is recommended. As a pathophysiology of NS, functional alterations in the RAS signaling pathway by gain of function mutation have been suggested.88) A patient-derived induced pluripotent stem cell-derived cardiomyocyte model was constructed to delineate the mechanism of RAF1-associated Noonan syndrome and HCM.89) Hyperactivation of mitogen-activated protein kinase kinase 1/2 (MEK 1/2) caused myofibrillar disarray and HCM. Because the RAS/MAPK signaling pathway plays a central role in cellular proliferation and differentiation, further studies related to HCM are expected.
Go to :
HISTOLOGICAL FEATURES OF HYPERTROPHIED MYOCARDIUM IN INFANTILE HYPERTROPHIC CARDIOMYOPATHY
A thick ventricular myocardium in HCM is due to addition of myofibrils in parallel, and intracellular myofibrillar disarray and intercellular disorganization.7) As mentioned above, proliferation of cardiomyocytes decreases between birth and 20 years old in humans because post-mitotic cardiomyocytes do not increase in number.15) Adult HCM shows an increased size of cardiomyocytes, disorganized arrangement, an increased amount of interstitial fibrosis, and diastolic dysfunction. Asymmetric ventricular wall hypertrophy is frequently observed in pathological specimens with an irregular direction of the myofibril array. However, infantile HCM cases frequently show diffuse ventricular myocardial hypertrophy.
An autopsy specimen of a 51-day-old infant with fulminant HCM showed myofibrillar hypertrophy with nuclear enlargement and hyperchromasia without prominent fibrosis. The prominent nuclei show a typical box shape in massively hypertrophied ventricular myocardium (Figure 3). Nuclear hypertrophy of cardiomyocytes in the human hypertrophic heart is characterized by a bizarre shape and clumping of chromatin. This reflects an increased biosynthetic activity of DNA repair, synthesis, transcription, and translational efficacy.90) Histologically, hereditary HCM cases are considered as pathological myofibrillar proliferation compared with apoptosis during hypertrophy of the myocardium. A regenerative response in neonatal mice after partial resection of cardiac tissue is characterized by proliferation of cardiomyocytes with minimal fibrosis, which is in contrast to 7-day-old or older mice that fail to regenerate after resection.14) A murine model of myocardial infarction in 1-day-old mice by coronary arterial ligation showed complete cardiac regeneration without scarring 7 days after an ischemic insult.91) The majority of cardiomyocytes within regenerated tissue originate from preexisting cardiomyocytes in neonates.14) Dedifferentiation of regenerating cardiomyocytes have marked sarcomere disassembly in response to heart injury.92)
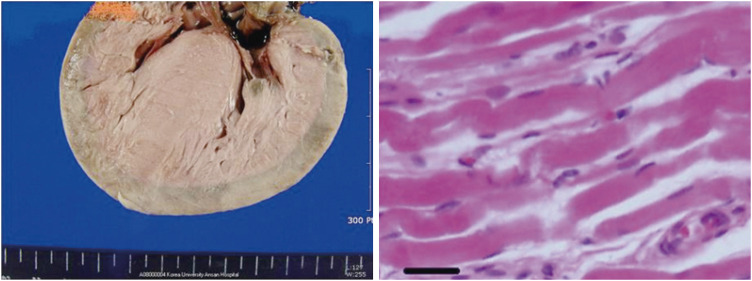 | Figure 3Gross specimen of fatal fulminant infantile HCM (left) shows diffuse hypertrophy of the ventricular wall without prominent fibrosis. Microscopic findings of the myocardium (right) show hypertrophy of cardiomyocytes with nuclear enlargement and hyperchromasia. The nuclei predominantly show a typical box shape (bar = 50 μm) (reproduced with permission from Seo HS, et al. Korean Circ J 2013;43:54–6).HCM = hypertrophic cardiomyopathy.
|
In clinical practice, infantile HCM shows diffuse LV wall hypertrophy with less fibrosis compared with late-onset HCM in adults in cardiac imaging studies. A histopathological study of postmortem myocardial biopsies of young patients with HCM showed a statistically significant difference in scar-type fibrosis in terms of mean age, mean septal thickness and there was no septal fibrosis in young children (1, 4, 5, and 12 years old).93) Moreover, especially extensive septal replacement-type fibrosis was exclusive of asymmetric HCM.93)
With regard to the pattern of fibrosis in dilated cardiomyopathy, patchy, diffuse, and perivascular fibrosis differs between pediatric and adult patients. Patchy fibrosis is frequently found in pediatric patients, whereas perivascular fibrosis is predominantly found in older hypertensive adults.94) A neonate who was diagnosed with HCM and NS underwent cardiac transplantation at the age of 36 weeks.95) Endomyocardial biopsy at 12 weeks showed mild perimuscular interstitial fibrosis with mild disarray of hypertrophic cardiomyocytes, with marked perivascular fibrosis and arteriolar wall thickening at the explanted heart at 36 weeks. Another case report documented two neonates of a mother with DM with HCM who died within 48 hours of birth from cardiorespiratory failure.96) This report showed that both neonates had hearts that were approximately twice the normal weight. However, there were only nonspecific findings, including some vacuolar and hydropic changes without disorganization of myocardial fibers in a histological examination.
Recent enthusiastic effort toward regenerative cardiology to repair injured myocardium has had success. However, there are still challenges and limitations of various regenerative strategies to treat the injured myocardium.97) Updated understanding of the proliferative nature of immature cardiomyocytes during the perinatal window may be helpful for regeneration of cardiomyocytes. In addition, studying the perspective of maturation of cardiomyocytes could add meaningful steps to understand infantile HCM (Figure 4).
 | Figure 4Proliferative hyperplastic immature cardiomyocytes versus non-proliferative hypertrophic postnatal mature cardiomyocytes and phenotypes of infantile HCM and late-onset HCM.HCM = hypertrophic cardiomyopathy.
*Footnote areas are referred from Bergmann et al.10)
|
Go to :
CONCLUSIONS
Fetal gene expression in heart failure in adults has been the focus of attention because of the limited capability of regeneration of cardiomyocytes after birth. Interestingly, rapid progression of ventricular hypertrophy in pediatric HCM shows two peaks during the infantile and adolescent periods that are roughly correlated with children's growth spurt. Recent advancement of molecular biology has increased our understanding of cardiac development from various points of view. During the perinatal period, hyperplastic immature cardiomyocytes transit to hypertrophic mature cardiomyocytes. This period corresponds to diverse postnatal metabolic and hormonal changes that are vital for survival after birth, and mechanotransductive stress during the transitional circulation affects maturation of immature cardiomyocytes. Pathological conditions, such as hypoxia, during the transitional circulation, abnormal hormonal changes, and premature birth comorbid with respiratory problems might inhibit the physiological transition of maturation of cardiomyocytes. This article reviews recent molecular and clinical studies to determine the peculiar characteristics of fulminant phenotypic presentation of infantile HCM from the perspective of maturation of cardiomyocytes. We summarized major characteristics of immature and mature cardiomyocytes in Table 1 as we described in this review. We expect better understanding of this condition with further basic researches on the exact mechanisms of infantile HCM in the future.
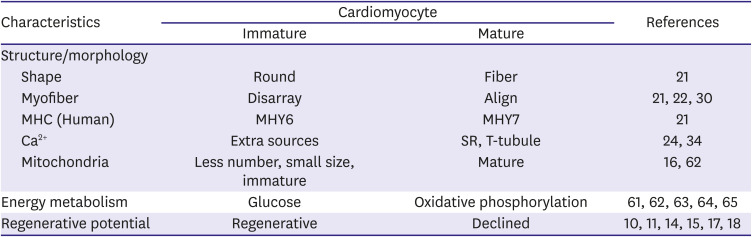
Table 1
Summary of major characteristics of immature and mature cardiomyocyte
| Characteristics | Cardiomyocyte | References | ||
|---|---|---|---|---|
| Immature | Mature | |||
| Structure/morphology | ||||
| Shape | Round | Fiber | 21 | |
| Myofiber | Disarray | Align | 21 22 30 | |
| MHC (Human) | MHY6 | MHY7 | 21 | |
| Ca2+ | Extra sources | SR, T-tubule | 24 34 | |
| Mitochondria | Less number, small size, immature | Mature | 16 62 | |
| Energy metabolism | Glucose | Oxidative phosphorylation | 61 62 63 64 65 | |
| Regenerative potential | Regenerative | Declined | 10 11 14 15 17 18 | |
Characteristics classified by structure, energy and regeneration between immature and mature cardiomyocytes are summarized with references.
MHC = myosin heavy chain; SR = sarcoplasmic reticulum; T-tubule = transverse tubule.
![]()
Ethical statement
This study was approved by the Institutional Review Board of St. Vincent's Hospital, The Catholic University of Korea (approval No. VC21ZASI0068).
Go to :
 XML Download
XML Download