

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Cisplatin is a highly effective anticancer drug used to treat patients with bladder, head, neck, lung, ovarian, and testicular cancers [1]. The most common adverse effects of cisplatin treatment in patients with cancer are muscle weakness and fatigue due to skeletal muscle mass depletion [2]. Specific muscular atrophy induced by cisplatin treatment is associated with autophagy activation. Previous studies showed that the forkhead box O3 (FOXO3a) transcription factor was retained in the nucleus following dephosphorylation of Thr32 and Ser253, leading to promotion of the transcription of autophagy-related factors including lipidated microtubule-associated protein 1 light chain 3 (LC3), p62, and Beclin 1 and activation of muscle atrophy through muscle RING finger protein 1 (MuRF 1) and Atrogin-1, which are involved in muscle wasting [3-5]. Particularly, Atrogin-1 and MuRF 1 accelerate skeletal muscle atrophy following cisplatin treatment in human and animals [1,6-8].
In animal models, cisplatin treatment was shown to stimulate cell autophagy in fast-twitch skeletal muscle by downregulating the Akt and phosphorylated FOXO3a signaling pathways [9,10]. Combining exercise with cisplatin treatment can prevent muscle atrophy by altering the autophagy signaling pathway. Exercise is also related to the expression of AMP-activated protein kinase-regulated skeletal muscle protein metabolism as well as increased levels of autophagy-related FOXO3a during cisplatin treatment [11]. Exercise-preserved muscle mass and function during cisplatin treatment was shown to be associated with the regulation of autophagy [12]. In previous study, aerobic exercise performed during cisplatin treatment attenuated body weight loss by 50% and maintained lean body mass and muscle grip strength [13]. In addition, seven weeks of aerobic exercise protected against cell death in C57BL6 male mice treated with a single injection of cisplatin [14], and cisplatin administered to fast-twitch skeletal muscle affected autophagy by decreasing the levels of protein kinase B (AKT) and phosphorylated FOXO3a [15]. However, the impact of different exercise types (e.g., aerobic and resistance exercise) on autophagy and muscle atrophy factors, particularly on the levels of AKT, peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), and FOXO3a, in different muscle fiber types (e.g., type I and type II muscle) is not well-understood.
In clinical studies, mice are typically treated with 1 mg/kg cisplatin, which affects levels of autophagy factors [16,17]. Previous studies showed that aerobic exercise prevents muscle atrophy; however, different exercise types and the key mediator autophagy factor FOXO3a have not been evaluated in different skeletal muscle types [1,18,19]. Exercise has been shown to increase PGC-1α and AKT, suggesting that exercise can potently regulate autophagy-related factors in cisplatin-induced muscle wasting [20,21]. Therefore, this study was conducted to evaluate the effect of two different exercise types—aerobic and resistance exercise—on the expression levels of various autophagy-related factors, including FOXO3a, Beclin 1, BCL2-interacting protein 3 (BNIP3), and the LC3-II/I ratio, as well as on muscle atrophy. Atrophy was assessed by measuring the expression levels of 4EBP1, MuRF 1, and Atrogin-1 in rats treated with the current standard cisplatin dosage. We hypothesized that different aerobic and resistance exercises would decrease the expression levels of autophagy-related factors and muscle atrophy following cisplatin treatment.
Go to : 
METHODS
Animal care and experimental protocols
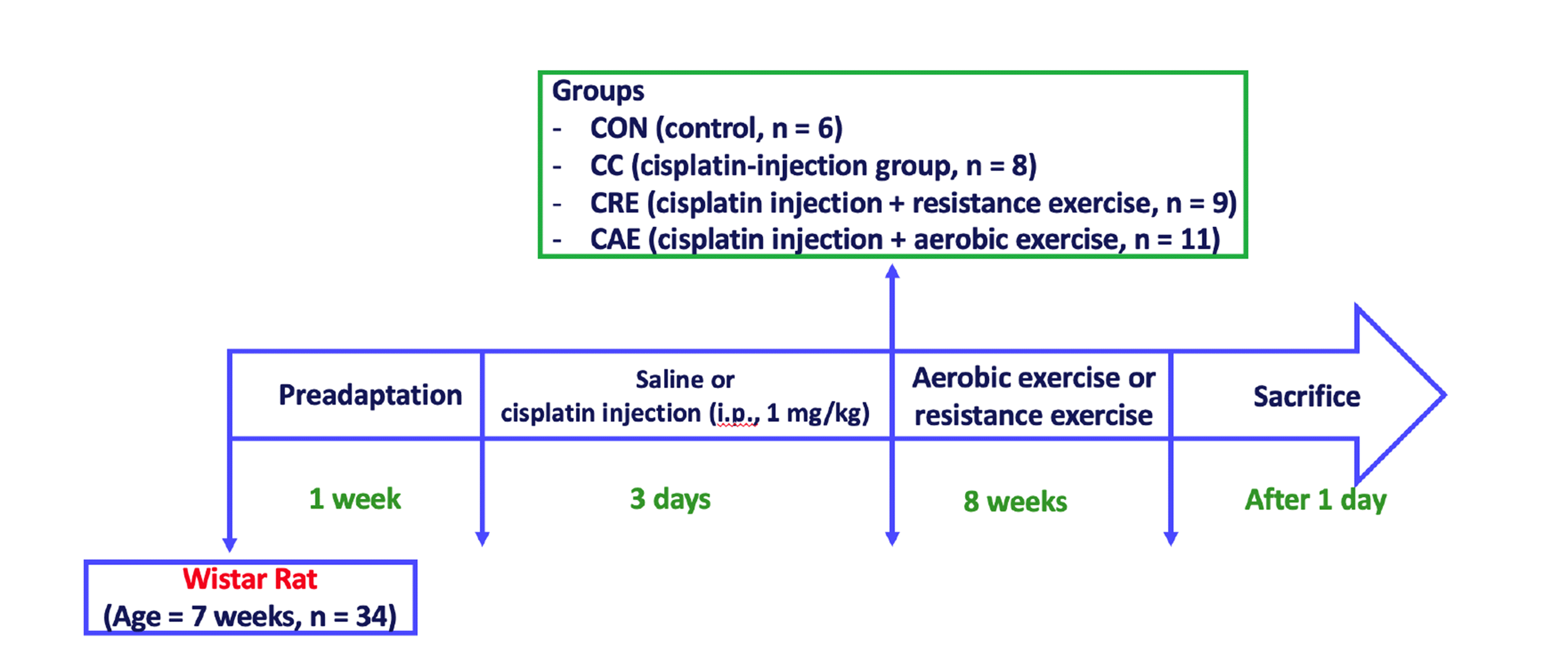
All experiments were approved by the Institutional Animal Use and Care Committee of Inje University (Busan, Korea: IACUC approval No. 2018-013). Seven-week-old male Wistar rats (n = 34) were purchased from Orient Bio (Seongnam, Korea) and housed at a temperature of 23°C and humidity of 40%–60%, with a 12:12-h light–dark cycle. The rats were fed a standard diet and water ad libitum. After one week of acclimatization, the rats were randomly assigned to one of four groups subjected to different treatments and/or exercise regimes: control group (CON, n = 6), administered only saline injections; CC group (n = 8), administered cisplatin injections (1 mg/kg) without exercise; CRE group (n = 9), administered cisplatin (1 mg/kg) + resistance exercise; CAE group (n = 11), administered cisplatin (1 mg/kg) + aerobic exercise. The animals were administered cisplatin (1 mg/kg) once daily for three days [15] and allowed to adapt to their environment for one week prior beginning aerobic exercise and resistance training. During this week, rats in the CAE group were familiarized with treadmill running (1050-RM, Exer-3/6 treadmill; Columbus Instruments, Columbus, OH, USA) four times per week at a pace of 20 m/min for 15 min. During the first week after familiarization, the rats exercised by treadmill running at 10 m/min on a 0% gradient for 10 min/day. The exercise intensity was gradually increased to 20 m/min on a 0% gradient for 60 min/day over eight weeks [22]. During the week following the three-day treatment regime, rats in the CRE group were placed on a 1-m ladder with a 2-cm interval grid at an 85° angle five times per week. Resistance exercise training was progressively increased by 10% of the body weight each week [23] for 60 min/day over eight weeks. All rats were euthanized by intraperitoneal injection of alfaxalone and then dissected. The experimental procedure is illustrated in Fig. 1. The gastrocnemius (GAS) and soleus (SOL) muscle tissues were collected and frozen in liquid nitrogen for storage at –80°C until analysis.
Western blotting
We extracted total protein from the GAS and SOL muscle tissues using RIPA buffer (#89900; Thermo Fisher Scientific, Waltham, MA, USA) containing phosphatase inhibitor (#4906845001; Sigma-Aldrich, St. Louis, MO, USA) and protease inhibitor (#4693159001; Roche, Basel, Switzerland). Proteins were separated using 10%–16% Tris-glycine sodium dodecyl sulfate–polyacrylamide gel electrophoresis and then transferred onto nitrocellulose membranes using iBlot 2 Transfer Stacks (#IB23002; Invitrogen, Carlsbad, CA, USA). The following primary antibodies were used: LC3 (#L7543) and p62 (126M4796V) from Sigma-Aldrich; BNIP3 (#3769), Beclin 1 (#3738), FOXO3a (#2497), p-FOXO3a (#8174), AKT (#4691), p-AKT (#9271), mTOR (#2972), p-mTOR (#5536), 4EBP1 (#9644), and p-4EBP1 (#9459) from Cell Signaling Technology (Danvers, MA, USA); and PGC1-α (#SC-518025; Santa Cruz Biotechnology, Dallas, TX, USA), MuRF 1 (#EPR6431(2); Abcam, Cambridge, UK), and Atrogin-1 (#AP2041; ECM-Biosciences, Versailles, KY, USA). Each primary antibody was diluted by 1:1,000 with Tris-buffered saline containing Tween 20 (#HT2007; Biosesang, Seongnam, Korea) containing 5% skim milk at 4°C overnight. The nitrocellulose membranes were then incubated with a peroxidase-conjugated secondary anti-rabbit antibody (#7074; Cell Signaling Technology), and specific antibodies were detected using Immobilon Western Chemiluminescent HRP Substrate (#WBKLS0500; MilliporeSigma, Billerica, MA, USA). The band densities were normalized to that of glyceraldehyde-3-phosphate dehydrogenase (GAPDH), which was used as an internal control.
Statistical analyses
All data are presented as the mean ± standard deviation. Statistical analyses were performed using GraphPad Prism version 9.1.0. software (GraphPad, Inc., San Diego, CA, USA). Different groups were evaluated using one-way analysis of variance and Tukey’s honestly significant difference test for post-hoc analysis. A p-value of ≤ 0.05 was considered to indicate a statistically significant difference.
Go to :
RESULTS
Exercise improved autophagy-related protein levels in skeletal muscle
Western blot analysis of the GAS muscle samples (Fig. 2) from the CRE and CAE groups revealed lower levels of the autophagy-related proteins Beclin 1 (Fig. 2B: F = 148.40, p < 0.001), p62 (Fig. 2D: F = 18.20, p < 0.001), and BNIP3 (Fig. 2E: F = 85.83, p < 0.001) and a lower LC3-II/I ratio (Fig. 2F: F = 36.32, p < 0.001) compared to in the CC group. In addition, FOXO3a was significantly upregulated in both the CRE and CAE groups (Fig. 2C: F = 69.06, p < 0.001). Western blot analysis of the SOL muscle samples (Fig. 3) showed that Beclin 1 levels were downregulated (Fig. 3B: F = 78.27, p < 0.001), the LC3-II/I ratio was decreased (Fig. 3F: F = 28.21, p < 0.001), and p62 levels were downregulated (Fig. 3D: F = 11.42, p < 0.001) in both the CRE and CAE groups, whereas FOXO3a was upregulated in these groups (Fig. 3C: F = 37.49, p < 0.001). BNIP3 (Fig. 3E: F = 22.05, p < 0.001) levels in the SOL muscle samples were upregulated in the CRE group but did not differ significantly from those in the CC group.
 | Fig. 2Expression levels of autophagy-related factors in gastrocnemius muscle.(A, G) Representative bands. (B) Beclin 1/GAPDH, (C) phospho-FOXO3a/total-FOXO3a, (D) p62/GAPDH, (E) BNIP3/GAPDH, (F) LC3-II/I ratio, (H) phospho-mTOR/total-mTOR, (I) phospho-Akt/total-Akt, and (J) PGC1-α/GAPDH. CON, control (n = 6); CC, cisplatin control (n = 8); CRE, resistance exercise with cisplatin treatment (n = 9); CAE, aerobic exercise with cisplatin treatment (n = 11). AKT, protein kinase B; FOXO3a, forkhead box O3; PGC1-α, peroxisome proliferator-activated receptor gamma coactivator 1-alpha; LC3, lipidated microtubule-associated protein 1 light chain 3; p62, sequestosome-1; Beclin 1, autophagy-related 6 homolog; BNIP3, BCL2-interacting protein 3; GAPDH, glyceraldehyde-3-phosphate dehydrogenase. One-way analysis of variance and Tukey’s post-hoc were used for statistical analysis; units are arbitrary. *p < 0.05, **p < 0.01, ***p < 0.001.
|
 | Fig. 3Expression levels of autophagy-related factors in the soleus muscle.(A, G) Representative bands. (B) Beclin 1/GAPDH, (C) phospho-FOXO3a/total-FOXO3a, (D) p62/GAPDH, (E) BNIP3/GAPDH, (F) LC3-II/I ratio, (H) phospho-mTOR/total-mTOR, (I) phospho-Akt/total-Akt, and (J) PGC1-α/GAPDH. CON, control (n = 6); CC, cisplatin control (n = 8); CRE, resistance exercise with cisplatin treatment (n = 9); CAE, aerobic exercise with cisplatin treatment (n = 11). AKT, protein kinase B; FOXO3a, forkhead box O3; PGC1-α, peroxisome proliferator-activated receptor gamma coactivator 1-alpha; LC3, lipidated microtubule-associated protein 1 light chain 3; p62, sequestosome-1; Beclin 1, autophagy-related 6 homolog; BNIP3, BCL2-interacting protein 3; GAPDH, glyceraldehyde-3-phosphate dehydrogenase. One-way analysis of variance and Tukey’s post-hoc were used for statistical analysis; units are arbitrary. *p < 0.05, **p < 0.01, ***p < 0.001.
|
Exercise increased AKT, mTOR, and PGC1-α levels in skeletal muscle of cisplatin-induced rats
The phosphorylation of AKT (Fig. 2I: F = 13.03, p < 0.001), mTOR (Fig. 2H: F = 87.22, p < 0.001), and PGC1-α (Fig. 2J: F = 298.90, p < 0.001) showed a greater increase in the GAS muscle samples from the CRE and CAE groups (Fig. 2) than in those from the CON and CC groups. In the SOL muscle (Fig. 3), phosphorylated mTOR showed greater upregulation only in the CRE group compared to in the CON group (Fig. 3H: F = 26.78, p < 0.001). In addition, PGC1-α (Fig. 3I: F = 119.10, p < 0.001) exhibited greater upregulation in the CAE and CRE groups than in the CC and CON groups. Based on these results, exercise interventions may increase mitochondrial biogenesis in cisplatin-induced rats.
Exercise regulated AKT/PGC1-α/FOXO3a signaling pathways in skeletal muscle of cisplatin-induced rats
We investigated the effects of PGC1-α-regulated FOXO3a activation on the skeletal muscle of cisplatin-induced rats. In both the GAS (Fig. 2) and SOL (Fig. 3) muscles subjected to cisplatin treatment, the AKT/FOXO3a signaling pathways were modulated, which can result in increased autophagy. The p-FOXO3a/FOXO3a ratios in the cisplatin-induced skeletal muscle of the CC group were significantly decreased. Thus, both aerobic and resistance exercise improved the deficiency of autophagy-related factors in cisplatin-treated skeletal muscle by regulating the AKT/PGC1-α/FOXO3a signaling pathway.
AKT/PGC1-α/FOXO3a signaling pathway directly affected 4EBP1, MuRF 1, and Atrogin-1 levels following exercise training
As shown in Fig. 4, the GAS (Fig. 4B: F = 32.16, p < 0.001) and SOL (Fig. 4F: F = 20.41, p < 0.001) muscle samples from the CRE and CAE groups showed significantly upregulated levels of phosphorylated 4EBP1 compared to in the CC group. Furthermore, both the GAS (Fig. 4D: F = 19.19, p < 0.001) and SOL (Fig. 4H: F = 19.17, p < 0.001) muscle exhibited higher upregulation of MuRF 1 and Atrogin-1 compared to that in the GAS (Fig. 4C: F = 6.06, p < 0.01) and SOL muscle (Fig. 4G: F = 47.85, p < 0.001) in the CRE and CAE groups. Therefore, muscle protein synthesis increased via upregulation of 4EBP1 following exercise, leading to decreased muscle atrophy.
 | Fig. 4Expression levels of muscle atrophy-related proteins including 4EBP1, Atrogin-1, and MuRF 1, in the gastrocnemius and soleus muscles.(A, E) Representative bands. (B) Phospho-4EBP1/total 4EBP1 in gastrocnemius, (C) Atrogin-1/GAPDH in gastrocnemius, (D) MuRF 1/GAPDH in gastrocnemius, (F) phospho-4EBP1/total 4EBP1 in soleus, (G) Atrogin-1/GAPDH in soleus, and (H) MuRF 1/GAPDH in soleus. CON, control (n = 6); CC, cisplatin control (n = 8); CRE, resistance exercise with cisplatin treatment (n = 9); CAE, aerobic exercise with cisplatin treatment (n = 11). MuRF 1, muscle RING finger protein 1; Atrogin-1, muscle-specific F-box protein; GAPDH, glyceraldehyde-3-phosphate dehydrogenase. One-way analysis of variance and Tukey’s post-hoc were used for statistical analysis; units are arbitrary. *p < 0.05, **p < 0.01, ***p < 0.001.
|
Go to :
DISCUSSION
We examined the role of autophagy-related factors during cisplatin administration combined with exercise and found that both resistance and aerobic exercise improved the autophagy-related factor expression and reduced muscle atrophy, with no apparent differences between the exercise types.
In cisplatin-treated GAS muscle, the Beclin 1, p62, and BNIP3 levels were downregulated and LC3-II/I ratio was decreased in the CRE group compared to in the CC group (Fig. 2B, D–F). These factors were also downregulated in the CAE group relative to in the CC group, except for p62 (Fig. 2B, E, F). Moreover, phosphorylation of FOXO3a in the GAS and SOL muscles following cisplatin treatment was upregulated in both the CRE and CAE groups (Figs. 2C and 3C). These results are supported by the upregulation of Beclin 1, LC3-II, and p62, and FOXO3a has been shown to transcriptionally activate autophagy and several autophagy-related genes as well as LC3 and Beclin 1 in a cisplatin-treated model [15,24,25]. Our results also showed that exercise modulated the expression of autophagy-related factors and may be involved in the adaptation of autophagy-related factors in the muscle and increased levels of oxidative proteins [26].
Our findings were supported by those of a previous study showing that 6 weeks of aerobic exercise did not affect skeletal muscular inflammation and glucose tolerance but preserved muscle mass in cisplatin-administered mice [13]. In addition, in a cisplatin-administered mouse model, phosphorylation of AKT and FOXO3a was increased and myostatin (Mstn) gene expression was attenuated in the quadriceps and gastrocnemius [27]. Another previous study indicated that only aerobic exercise can protect against muscle atrophy by altering the levels of MuRF 1 and Atrogin-1; however, our findings indicate that resistance exercise in cisplatin-administered rats did not dramatically affect MuRF 1 and Atrogin-1 levels compared with aerobic exercise training. This may be because aerobic exercise training is more reliant on degradation of the ubiquitin proteasome pathway marker FOXO3a compared to in resistance training [28].
We found that AKT, FOXO3a, and PGC1-α expression levels were higher in the GAS and SOL muscles following cisplatin treatment in the CRE and CAE groups than in the CC group (Fig. 2C, I, J; Fig. 3C, I, J). A previous study showed that the AKT/PGC1-α/FOXO3a signaling pathway decreased autophagy-related factors, including BNIP3, LC3, Beclin 1, and p62 [1], during cisplatin treatment. We found that the level of phosphorylation of AKT, PGC1-α, and FOXO3a was increased by both resistance and aerobic training, leading to downregulation of BNIP3, Beclin 1, and p62 and a decrease in the LC3-II/I ratio. PGC1-α regulates autophagy [29], and exercise-induced AMP-activated protein kinase decreases the regulation of FOXO3a, which can in turn decrease the transcription levels of LC3, p62, and BNIP3 [30-33]. These findings also suggest that increased PGC1-α levels help prevent autophagy in the skeletal muscles caused by cisplatin-induced cytotoxicity [20]. A previous study demonstrated that patients with cancer treated with chemotherapy showed no changes in the skeletal fiber type distribution after 10 weeks of exercise [34], which agrees with our results, revealing similar responses in the expression levels of AKT, PGC1-α, and FOXO3a in the GAS and SOL muscles. Our findings indicate that exercise training directly affects the phosphorylation of FOXO3a and overexpression of PGC1-α to modulate autophagy-related factors during cisplatin treatment.
In this study, muscle atrophy was prevented as demonstrated by the downregulation of Atrogin-1 and MuRF 1 in both the CRE and CAE groups (Fig. 4C, D, G, H). In addition, the level of phosphorylation of 4EBP1 in CRE and CAE was increased in both the GAS and SOL muscle samples (Fig. 2B, F). This result is supported by a previous study reporting downregulation of muscle atrophy-related genes, such as those encoding MuRF 1 and Atrogin-1, in the aerobic exercise group [13]. Phosphorylation of 4EBP1 during exercise protects against muscle atrophy by increasing protein synthesis [35,36]. MuRF 1 and Atrogin-1 expression levels were decreased in the CRE and CAE groups, similar to the decreased levels of the autophagy-related factors BNIP3, Beclin 1, and p62 and decrease in the LC3-II/I ratio. Moreover, in tumor-bearing mice, aerobic exercise decreased the mRNA expression level of E3 ligase and levels of autophagy-related proteins as well as improved muscle protein turnover [37-39]. The muscles of patients with cancer cachexia showed increase BNIP3 and LC3 protein levels, suggesting that autophagy can be modulated by using a therapeutic approach [40]. Additionally, both aerobic and resistance exercise-induced FOXO3a expression in patients with cancer undergoing chemotherapy helped maintain the regulation of MuRF 1 and Atrogin-1, which prevented muscle wasting [41]. These findings indicate that exercise directly downregulated FOXO3a expression and de-activated the transcription of genes related to autophagy and muscle atrophy, thereby preventing muscle wasting during cisplatin treatment [26,29-31].
Autophagy and muscle atrophy occurred at similar levels in the GAS and SOL muscles, which is concordant with the results of previous studies showing that chemotherapy treatments caused the loss of type I muscle fibers [34] with no significant change after exercise training [42]. Additionally, in patients with breast cancer subjected to both aerobic and resistance training, MuRF 1 expression was downregulated in both type I and type II muscle fibers [43]. Moreover, in an autophagy knockout model (Atg16L1 mouse), attenuation of ATG16 autophagy proteins was highly correlated with skeletal muscle fiber development [44]. Overall, both the GAS and SOL muscles decreased autophagy expression levels for both exercise and skeletal muscle types (Fig. 5).
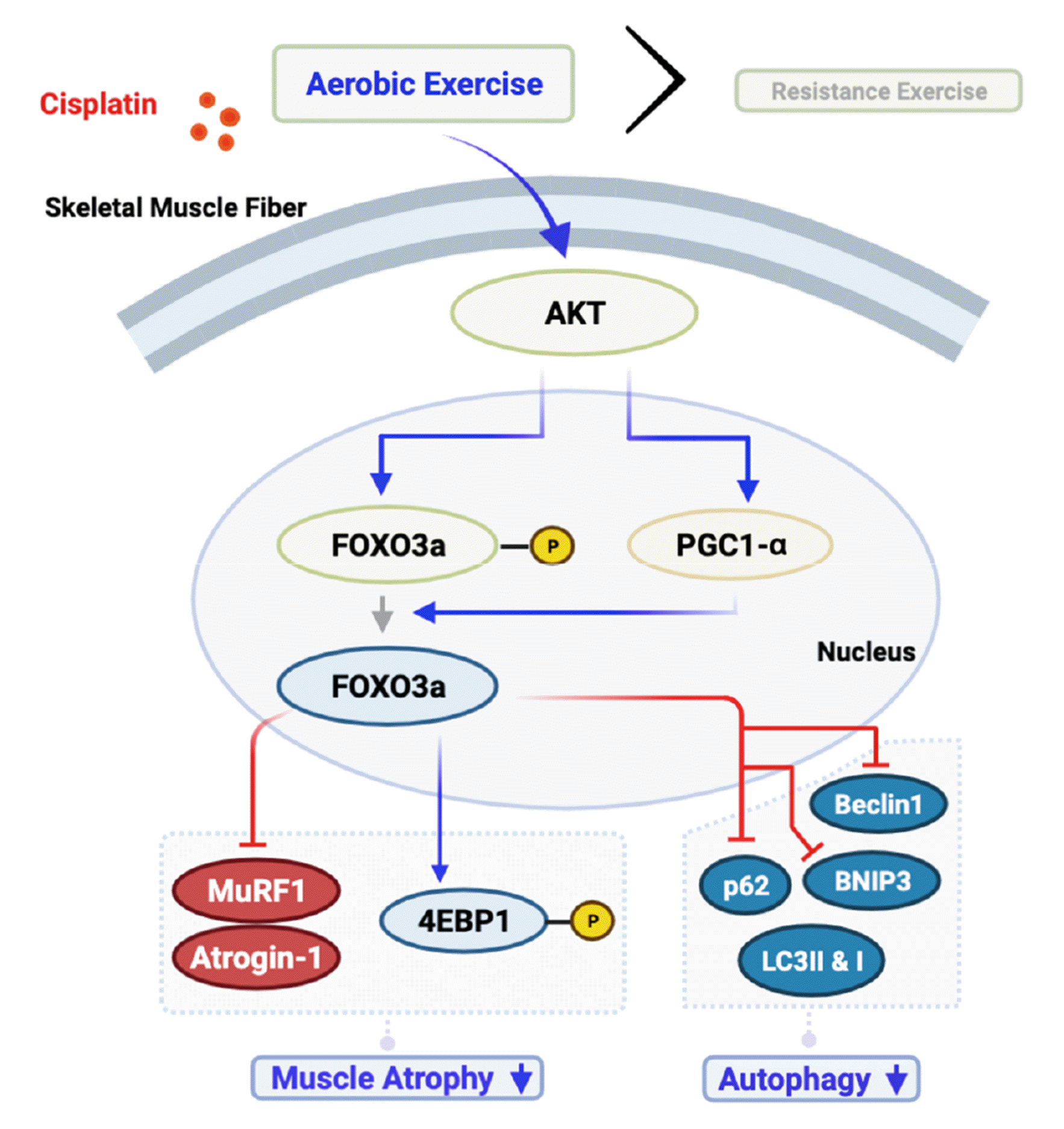 | Fig. 5AKT/PGC1-α/FOXO3a signaling pathways affected muscle atrophy and autophagy regulation.AKT, protein kinase B; FOXO3a, forkhead box O3; PGC1-α, peroxisome proliferator-activated receptor gamma coactivator 1-alpha; MuRF 1, muscle RING finger protein 1; Atrogin-1, muscle-specific F-box protein; LC3, lipidated microtubule-associated protein 1 light chain 3; p62, sequestosome-1; Beclin 1, autophagy-related 6 homolog; BNIP3, BCL2-interacting protein 3.
|
Exercise training directly affected the expression of AKT, PGC1-α, and FOXO3a, which modulated autophagy-related factors such as the expression levels of BNIP3, Beclin 1, and p62; the LC3-II/I ratio; and muscle atrophy, reflected by expression levels of MuRF 1 and Atrogin-1. These results suggest that both aerobic and resistance exercise inhibited muscle wasting by upregulating autophagy-related factors and downregulating muscle atrophy factors across different skeletal muscle types following cisplatin administration. Further studies are needed to analyze the effects of exercise on a cancer cachexia model and normal healthy controls.
Go to :
 XML Download
XML Download