

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Niemann-Pick disease type C (NPC, OMIM#257220, OMIM#607625) is an autosomal recessive lysosomal storage disorder caused by mutations in the NPC1 and NPC2 genes.1 These genes encode NPC1 and NPC2, which are proteins that mediate cholesterol transport in cells.2 Defective proteins cause the accumulation of lipids in lysosomes, such as unesterified cholesterol, sphingomyelin, and glycolipids.3
The clinical manifestations of NPC are wide-ranging and depend on the age of onset. NPC is classified into the pre/perinatal type (<2 months), early infantile type (2 months to 2 years), late infantile type (2-6 years), juvenile type (6-15 years), and adolescent/adult type (>15 years).1,3,4 Visceral symptoms, such as cholestasis, ascites, hepatosplenomegaly, and pulmonary infiltrates, are predominant in the pre/perinatal and early infantile types. Neurological symptoms include hypotonia, developmental delay, mental decline, gait disturbance, dysphagia, and seizure.4
Genetic analysis of the NPC1 and NPC2 genes provides a definitive diagnosis of NPC. Biomarkers, such as oxysterols, lyso-sphingomyelin, and lyso-sphingomyelin 509, can also be helpful in the diagnostic process.4
The estimated incidence of NPC is approximately one in 120,000 live births.3 To the best of the authors’ knowledge, there are only two reported cases of pre/perinatal-type or infantile-type NPC in Korea.5,6 This paper reports a case of perinatal- type NPC that presented with neonatal cholestasis and hepatosplenomegaly that was diagnosed using a neonatal cholestasis gene panel.
CASE REPORT
A Korean male was born at a gestational age of 37 + 3 weeks with a birth weight of 3.1 kg (25th-50th percentile). He was the first child of healthy parents, and his prenatal ultrasonography was normal. At 5 days of age, he was admitted to a tertiary medical center for jaundice. The laboratory results revealed unconjugated hyperbilirubinemia and elevated liver function tests: total bilirubin 22.1 mg/dL (ref. 0.2-1.5 mg/dL); direct bilirubin 4.3 mg/dL (ref. 0-0.5 mg/dL); AST 66 U/L (ref. 1-40 U/L); and ALT 15 U/L (ref. 1-40 U/L). Abdominal ultrasonography and a hepatobiliary scan ruled out the possibility of biliary atresia. He received oral ursodeoxycholic acid (UDCA) and phototherapy because of the high unconjugated bilirubin level.
At 15 days of age, the infant was transferred to the Seoul National University Children's Hospital because of aggravated liver function abnormality. Upon arrival, his height, weight, and head circumference were 50 cm (25th-50th percentile), 3.3 kg (25th-50th percentile), and 34 cm (25th-50th percentile). A physical examination revealed hepatosplenomegaly with the liver and spleen, both three fingerbreadths palpable, while the neurological examination was normal. The initial laboratory values were as follows: white blood cell count 5,790/mm3; hemoglobin 11.0 g/dL; platelet count 111,000/mm3; total bilirubin 10.8 mg/dL; direct bilirubin 7.3 mg/dL; AST 334 U/L; ALT 85 U/L; GGT 46 U/L; cholesterol 194 mg/dL; albumin 2.9 g/dL (ref. 3.3-5.2 g/dL); bile acids 122.6 μmol/L (ref. 0-6.0 μmol/L); PT-INR 1.3; aPTT 54.9 sec; and ammonia 105 μg/dL (27.2-102.0 μg/dL). Chest radiography did not reveal pulmonary infiltrates or other abnormalities.
The viral studies, including HAV, HBV, HCV, toxoplasmosis, rubella, cytomegalovirus, herpes simplex virus, and Ebstein-Barr virus, conducted at 15 days of age, were negative. Metabolic screening, including neonatal screening tests, serum amino acid, and urine organic acid conducted at 17 days of age, revealed an increase in serum tyrosine (319 μmol/L [ref. 13-91 μmol/L]); however, urine succinylacetone was not detected. The other metabolic disease tests, including α1-antitrypsin and very-long-chain fatty acids, were normal. A neonatal cholestasis gene panel was tested at 20 days of age to check for underlying genetic causes. Abdomen ultrasonography revealed coarse echogenicity of the liver parenchyma with a globular appearance, a tortuous central portal vein with luminal dilatation, prominent hepatic arterial flow, and suspicious multifocal hypoechoic nodular liver lesions. A liver MRI, performed at 22 days of age revealed hepatosplenomegaly with parenchymal enhancement and periportal edema (Fig. 1). A liver biopsy, performed at 23 days of age revealed intracytoplasmic and canalicular cholestasis, periportal fibrosis, and bile duct proliferation (Fig. 2). Echocardiography and ultrasonography of the brain and spine were normal.
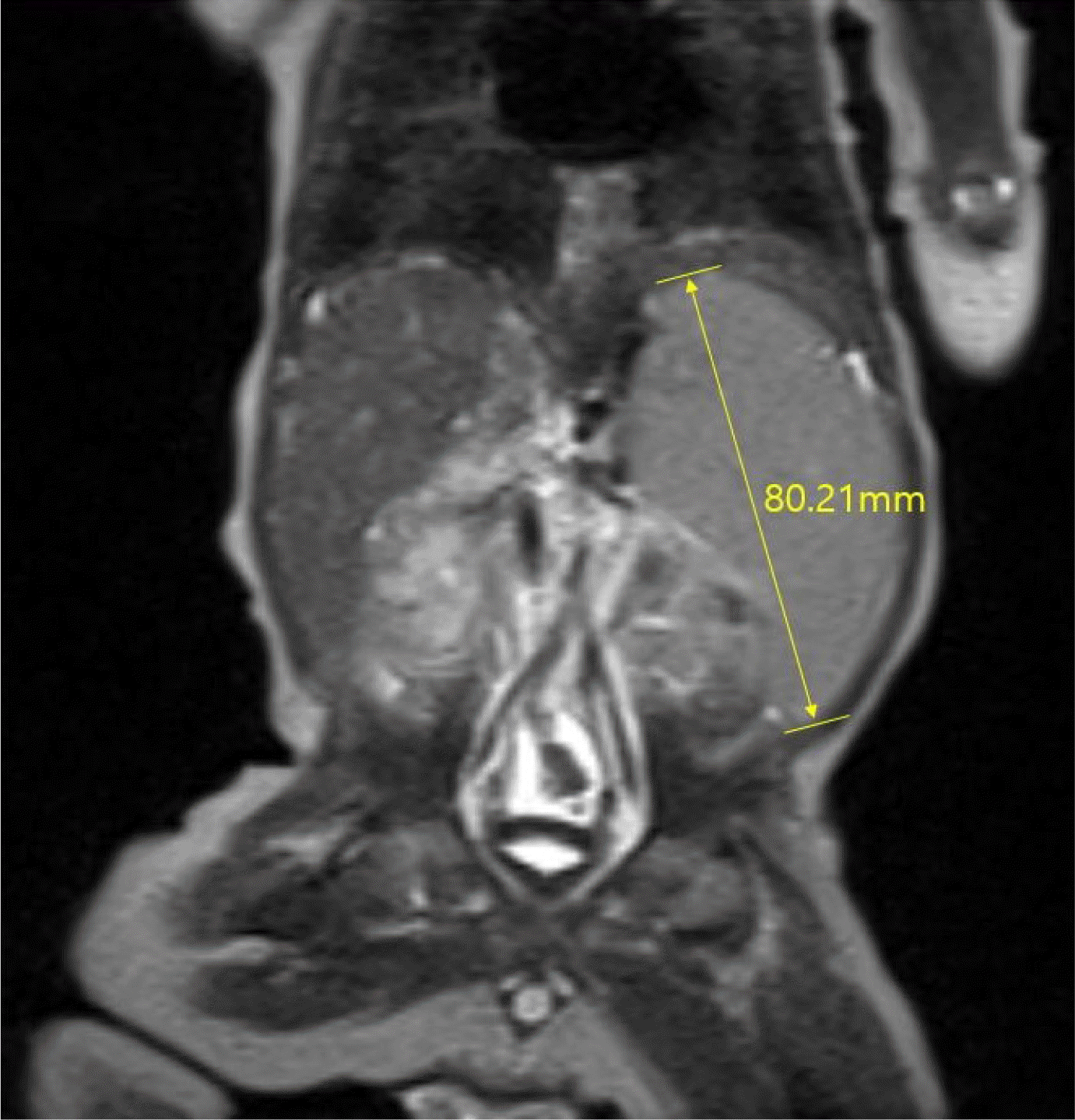
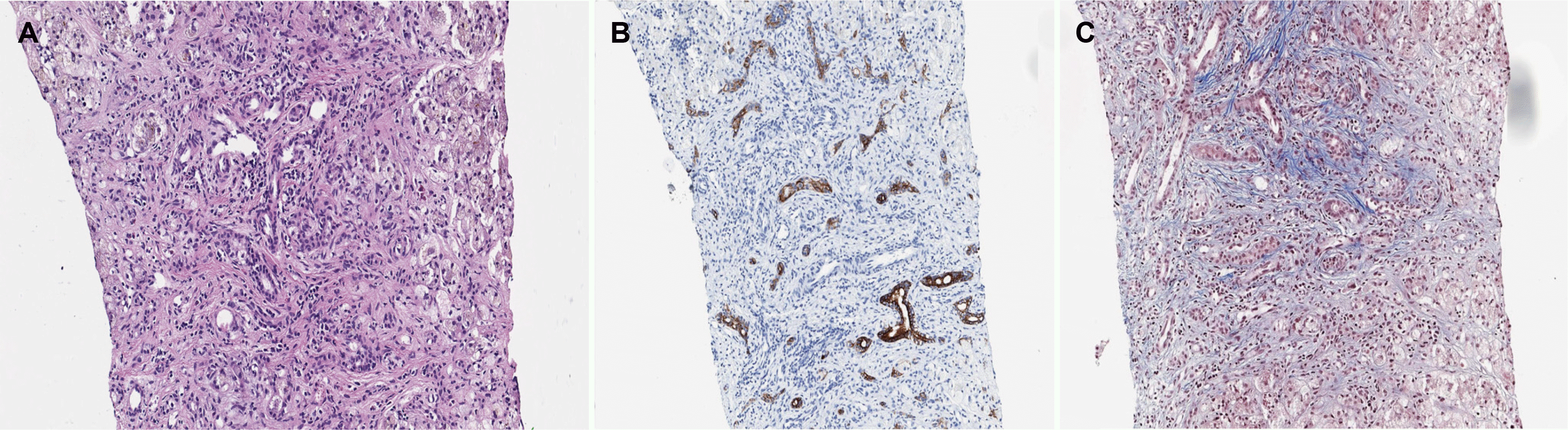
Fig. 1
Magnetic resonance imaging of the liver revealed hepatosplenomegaly, periportal edema, and a tortuous portal vein without obstruction.
![]()
Fig. 2
Liver biopsy: (A) H&E staining (×400) shows marked bile duct proliferation with intracytoplasmic and canalicular cholestasis. (B) Cytokeratin 7 (CK7) staining (×200) shows a moderate ductular reaction. (C) Masson’s trichrome (M-T) staining (×400) shows periportal and pericellular fibrosis, hepatocyte ballooning, and giant cell transformation.
![]()
The patient was provided with supportive management, including UDCA, fat-soluble vitamins, vitamin K, and diuretics. Oral sodium phenylbutyrate was given as the normal GGT levels suggested the possibility of progressive familial intrahepatic cholestasis type 2 (PFIC-2). Despite medical treatment, the ascites increased, and the liver function worsened, leading to liver failure. Abdominal circumference increased gradually over the course of admission from 35.5 cm to 44.0 cm. The laboratory results at 41 days of age were as follows: white blood cell count 9,110/mm3; hemoglobin 8.9 g/dL; platelet count 140,000/mm3; total bilirubin 23.4 mg/dL; direct bilirubin 17.7 mg/dL; AST 403 U/L; ALT 84 U/L; and PT-INR 1.5. A liver transplant was planned, but the parents did not want their child to undergo a surgical procedure that required a blood transfusion because of their religious beliefs. At 41 days of age, the patient was transferred to another tertiary medical center where bloodless surgery was available. Unfortunately, the patient died from gastrointestinal bleeding before the transplant could be performed.
The neonatal cholestasis gene panel, which took 12 weeks to provide results, revealed two novel likely pathogenic variants in the NPC1 gene: c.1145C>G (p.Ser382*) and c.2231_2233del (p.Val744del), leading to a diagnosis of NPC. The parents underwent genetic analysis of the NPC1, and both were carriers of each variant.
DISCUSSION
NPC is a progressive disease with several types depending on the age of onset. In the present patient, the onset of symptoms occurred at 5 days of age, putting him in the pre/perinatal- type NPC classification (<2 months). Pre/perinatal-type NPC manifests primarily as a liver disease with prolonged cholestasis, ascites, hepatosplenomegaly, and pulmonary infiltrates.4 In a previous report of seven patients with pre/perinatal- type NPC, splenomegaly was the most common finding, followed by fetal ascites.7 In most patients, the jaundice resolved spontaneously between 2 and 4 months. On the other hand, approximately 10% of patients with pre/perinatal-type NPC progress to acute liver failure, and their prognosis is generally poor, leading to death within 6 months.7,8 Although several countries have approved the use of miglustat for substrate reduction therapy to improve the neurological symptoms, it has not been shown to improve the visceral symptoms.9 In addition, a liver transplant is contraindicated for treating NPC.10 A recent case report from Japan presented three neonatal-onset NPC patients who underwent a liver transplant, with two diagnosed after the transplant and one diagnosed before.11 The transplant temporarily extended the lives of these patients with severe liver disease, but they ultimately experienced neurological deterioration.
More than 400 mutations in the NPC1 gene have been identified.12 A previous study observed a genotype-phenotype correlation after analyzing certain gene mutations and the corresponding biochemical data and classified the mutations as moderate or severe.13 The authors predicted that severe mutations, whether in the homozygous form or the compound heterozygous form, result in a severe phenotype. Because the present patient presented with a severe phenotype, the two novel mutations (c.1145C>G [p.Ser382*] and c.2231_2233del [p.Val744del]) in this patient are likely to be severe mutations.
The NPC patient presented with neonatal cholestasis. Neonatal cholestasis has more than 100 causes, including infections, toxins, endocrine disorders, anatomical problems, metabolic diseases, and genetic causes. Neonatal cholestasis is defined as hyperbilirubinemia found in infants or neonates, associated with direct bilirubin consisting of more than 20% of total bilirubin. Of the many causes of neonatal cholestasis, those that must be considered foremost are a TORCH infection and hepatitis, biliary atresia, and genetic diseases, including Alagille syndrome and citrin deficiency. The initial laboratory tests should include infection workup, including TORCH, HAV, HBV, and HCV, and metabolic screening. Liver ultrasonography is performed for an imaging evaluation. A liver biopsy should be considered if biliary atresia is not excluded by ultrasonography. If the common causes mentioned above are fully excluded, the neonatal cholestasis gene panel should be considered. Identifying the underlying cause of neonatal cholestasis is important because the timely treatment of a treatable cause can improve the prognosis. For example, infections can be treated with antimicrobials; galactosemia can be treated by changing the diet; biliary atresia can be treated via corrective surgery at an appropriate time.10
In addition to a routine evaluation of the liver enzymes and bilirubin levels, the GGT level can help determine the etiology of cholestasis because elevated GGT levels are usually associated with a biliary obstruction. A high level of GGT is indicative of conditions, such as biliary atresia, bile duct obstruction, neonatal sclerosing cholangitis, and PFIC type 3. A low or normal GGT level is not a common characteristic of neonatal cholestasis and is indicative of diseases, such as PFIC type 1, PFIC type 2, and bile acid synthesis disorders.10,14 In this patient, the GGT levels were within the normal range. Hence, for the initial differential diagnosis, diseases associated with biliary obstruction were less likely.
Recent advances in genetic testing have allowed the increased diagnosis of neonatal cholestasis with unidentified etiologies. In several studies, genetic analysis resulted in the molecular diagnosis of neonatal cholestasis in 22-27% of patients.15-17 Thus, the emerging approach to determining the cause of neonatal cholestasis is to exclude biliary atresia and other treatable conditions and then use a targeted gene panel or exome sequencing.10 The gene panel at the Seoul National University Children's Hospital was critical in the evaluation of the present patient. The institutional neonatal cholestasis panel included genes that cause the following diseases: PFIC, congenital bile acid synthesis defect, citrin deficiency, Rotor syndrome, Alagille syndrome, and Niemann–Pick disease (Supplementary Table 1). After the patient’s diagnosis, family screening revealed both parents to be carriers of the NPC1 gene mutation. Hence, genetic counseling should be provided when planning future pregnancies. Although the result of the neonatal cholestasis gene panel was not obtained before the patient’s death, his parents were thoroughly informed of the importance of early genetic counseling during future pregnancy to predict the genetic phenotype of the fetus before birth.
In conclusion, NPC must be considered in infants who present with neonatal cholestasis. In addition, the neonatal cholestasis gene panel may be crucial in the diagnostic process.
 XML Download
XML Download