

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Breast and ovarian cancers exert substantial social and financial burdens. Breast cancer was one of the most commonly diagnosed cancers in 2018 in the United States [1]. Every year, approximately 20,000 new cases of breast and ovarian cancers are detected in South Korea [2, 3]. The genetically well-characterized risk factors of breast and/or ovarian cancers include germline mutations of BRCA1 and BRCA2 genes. These genes are important tumor suppressors actively involved in the development of hereditary breast and ovarian cancer (HBOC) syndrome [4]. Up to 15% of germline mutations in the BRCA1 or BRCA2 gene are associated with the genetic risk factors of breast and ovarian cancers in the general population [5]. Deleterious germline mutations in either of the two genes may contribute up to 80% (by 70 years of age) lifetime risk of developing breast and ovarian cancer and are also related to early-onset disease [6-8]. BRCA1 and BRCA2 variants may be divided into three groups, namely, single-nucleotide variants or polymorphisms (SNVs or SNPs), small insertion/deletion (indels), and large genomic rearrangements [9].
The Sanger sequencing method is mainly used to detect mutations in BRCA1 and BRCA2 . However, it is time-consuming, labor-intensive, and expensive owing to the large size of BRCA1 and BRCA2 genes and the scattered mutations along these genes [10]. Next-generation sequencing (NGS) has been increasingly applied in cancer research and molecular diagnosis to simultaneously screen and intensively analyze different target genes [9]. This technique allows the genetic screening of BRCA1 and BRCA2 mutations to provide a high-throughput, quick, highly cost-effective, and comprehensive genome analysis.
We aimed to evaluate the clinical performance of BRCAaccuTest™ (NGeneBio, Seoul, Korea). We evaluated the analytical performance of the BRCAaccuTest™ kit as an NGS-based in vitro diagnostic (IVD) reagent. We also evaluated the ability of an NGS analytic software called NGeneAnalySys™ (NGeneBio) to automatically detect mutations in BRCA1 and BRCA2 genes and perform pathogenic classifications.
MATERIALS AND METHODS
1. Analytical validation of BRCAaccuTest™
To investigate the analytical performance of BRCAaccuTest™, control DNA samples were purchased from Coriell Institute (https://www.coriell.org/; See Supplemental Data Table S1). To test sensitivity (limit of detection), the input DNA was used at 50-, 10-, 5-, 1-, and 0.5-ng concentrations. Three reference DNAs, NA13713, NA14636, and NA14624, were used as positive controls of deleterious variants of either BRCA1 or BRCA2 . Different amounts (50, 10, 5, 1, and 0.5 ng) of each reference DNA were tested twice in separate experiments to assess the range of input gDNA. For the specificity test, healthy blood samples were supplemented with interfering substances such as bilirubin, hemoglobin, and cholesterol. Genomic DNA (gDNA) was extracted using a QIAGEN Blood DNA mini kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. In addition, the wash buffer from the DNA extraction kit was added as an interfering substance in the test sample (See Supplemental Data Table S2). To evaluate reproducibility, the experiment was carried out at two different locations, on different dates, with different batches of kits (different lot numbers), and/or performers. The input DNA was used at 10–50-ng concentrations, and two to three technical replicates were prepared for each test.
2. Sample selection for clinical test
The clinical samples used in this study were obtained from the Seoul National University Hospital (SNUH). Briefiy, gDNA was extracted from patients who underwent BRCA testing from 2008 to 2015. The previous BRCA test was applied to sequence BRCA1 and BRCA2 by the typical Sanger sequencing method at SNUH; the sequencing results were accumulated and well-documented by SNUH. Therefore, the leftover gDNA were identical to those used in the Sanger method and exploited to investigate the diagnostic consistency between the Sanger sequencing method and BRCAaccuTest™.
The residual gDNAs were maintained and controlled as per the regulations of the Ministry of Food and Drug Safety (MFDS) in the Republic of Korea and the Institutional Review Board (IRB) at SNUH. The samples satisfied the following criteria: 1) quantity was more than 100 ng for NGS or 5 μg for the Sanger method, 2) quality (A260/280) was 1.8–2.0, and 3) the storage period was less than 10 years. Samples were excluded from the study if they did not meet these criteria.
3. Clinical study design
This was a retrospective study performed by a single laboratory at SNUH under the regulation of MFDS in the Republic of Korea and IRB. It was a comparative study to assess the clinical utility and diagnostic consistency of BRCAaccuTest™ in comparison to the traditional Sanger sequencing method. As shown in Fig. 1, the target number of test samples was calculated based on the following criteria: diagnostic positive/negative agreement, 0.99; power, 80%; and statistical significance, 5% [11] with an additional 10% to compensate for drop-outs such as samples out of criteria. Therefore, the target number was initially set to 190 plus 22 extras for testing both BRCA1 and BRCA2 . Thus, the test samples for BRCA1 or BRCA2 were prepared with 106 samples each (total of 212 samples). Approximately 10% of the 212 test subjects (22 samples) were sequenced using the Sanger method to verify stability using an Applied Biosystems 3730xl DNA Analyzer (Applied Biosystems, Foster City, CA, USA) and a BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems) according to the manufacturer’s instructions. Ultimately, 207 samples met the calculated sample size (N=190).

Fig. 1
Design of the clinical study. The target number of test samples was determined according to the criteria of 0.99 diagnostic positive/negative agreement, 80% power, and 5% statistical significance. Abbreviations: PPA, positive percent agreement; NPA, negative percent agreement; OA, overall agreement.
![]()
4. NGS library preparation and sequencing
BRCAaccuTest™ was used to generate the NGS library of BRCA1 (NM_007294.3) and BRCA2 (NM_000059.3) genes according to the manufacturer’s instructions. Briefiy, the target regions of BRCA1 and BRCA2 genes were amplified using 10–50 ng of sample DNAs by the polymerase chain reaction (PCR). The amplicons were ligated using adaptors, and the library was amplified at the final step. Quantity and quality of the amplified library were evaluated using a Qubit® 3.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA) and Tapestation 2200 (Agilent Technologies, Santa Clara, CA, USA), respectively. After quality control (QC), the final library was normalized to a concentration of 4 nM and prepared for sequencing using an Illumina MiSeqDx® with MiSeq Reagent Nano Kit V2 (300 cycles) (Illumina, San Diego, CA, USA) as per the manufacturer’s instructions to generate pair-end reads.
5. Bioinformatic pipeline
The pipeline workfiow of data analysis was as follows: data QC, adapter trimming, alignment, variant calling, and annotation. Sequence reads generated were trimmed using Sickle v1.33 [12], a windowed adaptive trimming tool for sequencing adapters, and then aligned to the human hg19 reference genome using the BWA-MEM algorithm v0.7.10 [13]. Genetic variants, including SNVs and short insertions/deletions (indels), were identified using GATK v2.3 [14] and FreeBayes v9.9.2 [15], and the identified variants were annotated by snpEff v4.2 [16]. This bioinformatic pipeline was fully automated in NGeneAnalySys™ software. Sequence reads generated as FASTQ files could be uploaded on NGeneAnalySys™ to analyze, annotate, classify, and visualize NGS sequencing results, including clinical reports. Variants were classified according to the ACMG Standards and Guidelines for the Interpretation of Sequence Variants [17]. According to NGeneAnalySys™, the QC criteria for uniformity were set as follows: minimum coverage at >20× and average coverage at >200×.
6. Sanger sequencing
Peripheral blood samples were collected from family members of HBOC patients, non-familial breast cancer patients, or hereditary breast cancer patients. Genomic DNAs were extracted using the QIAGEN QIAamp DNA Blood Mini Kit (Qiagen) according to the manufacturer’s instructions. The residual genomic DNAs were stored at -70°C after Sanger sequencing for BRCA1 and BRCA2 . The BRCA1 and BRCA2 target regions were amplified using target-specific primers. The purified PCR products were sequenced using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems) according to the manufacturer’s instructions and analyzed using an Applied Biosystems 3730xl DNA Analyzer (Applied Biosystems).
7. Statistical analysis
The BRCA1 and BRCA2 alterations detected using BRCAaccuTest™ assay were compared with the results of the Sanger assay performed at SNUH. Positive percent agreement (PPA) and negative percent agreement (NPA) were defined as follows:
PPA=(BRCAaccuTestTM positive calls)/(Sanger assay positive calls)NPA=(BRCAaccuTestTM negative calls)/(Sanger assay negative calls)
To investigate the precision of BRCAaccuTest™, its reproducibility was tested by changing experiment performers, laboratories, dates, and lot numbers. The experiments for all combinations were carried out with three positive controls (NA13713, NA14636, and NA14624) and one negative control (NA12878). All samples were duplicated.
RESULTS
1. Analytic performance of BRCAaccuTest™
The analytical performance of BRCAaccuTest™ was tested to confirm its sensitivity, specificity, and precision. The test results demonstrated the successful production of all the sequenced libraries with an average 300× coverage depth in the target regions. Each representative BRCA mutation in the control DNA was also perfectly detected with 0.5 ng input DNA (Table 1). The NGS results illustrated no effects of the interfering substances on library preparation using BRCAaccuTest™ (Table 1). The precision of BRCAaccuTest™ showed 100% reproducibility in all combinations, namely, within-run, between-run, between-person, and between-lot (Table 1).
Table 1
Analytical performance of BRCAaccuTest™
![]()
2. Library preparation using BRCAaccuTest™
Approximately 10% of 212 test subjects (22 samples) were sequenced by the Sanger method to verify the stability of residual gDNA samples. All Sanger resequencing results were consistent with the previous ones. As the stability of the remaining samples was verified, subsequent NGS analysis was performed on 212 samples. In total, 207 samples (B001 to B207) were selected to create NGS libraries. These were divided into nine groups (runs) of 23 samples that were run on MiSeqDx® because the appropriate capacity of BRCAaccuTest™ is up to 24 samples, including 1 control (NA12878). The input DNA range of the 207 samples was 11.6–48.8 ng, and the average input DNA amount was 30.1±7.5 ng. The average time for library preparation for each group was 4.5–5.0 hours.
In total, 206 libraries of 207 samples were qualified and quantified at a success rate of 99.5%. Only one library, B021, was excluded owing to an insufficient amount of sample. As shown in Fig. 2A, all 206 libraries ranged from 15.2 to 100.3 nM, with an average of 55.2±15.0 nM (mean±SD). The BRCAaccuTest™ yielded 67.3 and 69.4 nM libraries with the input DNA of B15 (48.8 ng) and B101 (11.6 ng), respectively, indicating its highly consistent performance that was independent of input DNA amount. The average size of the final libraries, including the adapters, was 394.0±9.3 bp (Fig. 2B).
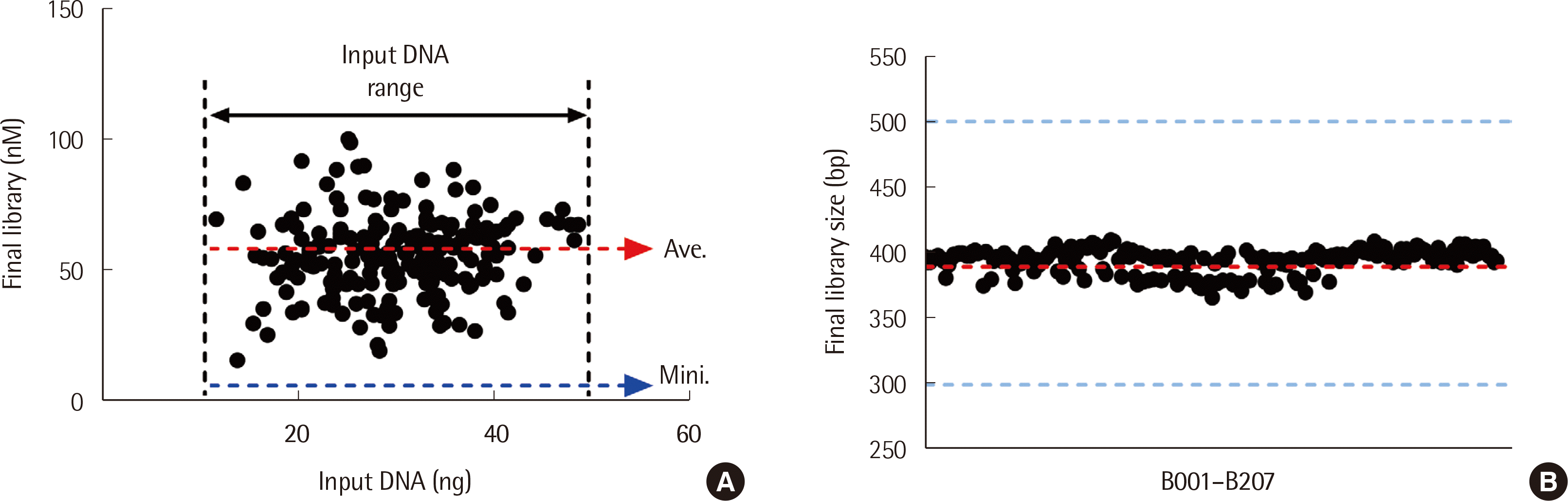
Fig. 2
Quantity and size of the final libraries. (A) The libraries of samples B001 to B207 were quantified and represented in nanomole (nM) quantities in correlation to the amount of input DNA (ng). The black dashed lines indicate the range of the input DNA used in this study. The red dashed arrow represents the average concentration of the library. The blue dashed arrow represents the minimum concentration of the library. (B) The size of each library was individually determined and represented as a dot. The red dashed line indicates the average size (bp) of the library. The blue dashed lines represent the range of the library size (300–500 bp). B021 was excluded owing to an insufficient amount of sample.
![]()
3. NGS
We successfully performed nine runs of NGS. Overall QC rejection rate was 4%. Only one sample, B022, showed a 13× minimum coverage at a QC failure rate of 0.04%. Therefore, there was no issue in analyzing the variants, as the minimum coverage region was irrelevant (data not shown). In addition, 95.6% of samples met the QC criteria of the average coverage.
4. Variant analysis
For variant analysis, ubiquitous homozygous SNPs in Asian populations, including c.4563A>G (rs206075), c.6513G>C (rs206076), and c.7397T>C (rs169547), found in BRCA2 were excluded (https://www.ncbi.nlm.nih.gov/clinvar/). A total of 1,704 variants were detected in 206 samples (including 8 samples as within-run and between-run intra-controls); 930 and 774 variants were detected in BRCA1 and BRCA2 , respectively. As 8 duplicated samples were excluded from the analysis, 1,640 variants were found in 198 samples. By excluding overlapped variants, 199 different types of variants (82 in BRCA1 and 117 in BRCA2 ) were estimated: 143 different SNVs (71.9%), 10 insertions (5.0%), 43 deletions (21.6%), and 3 indels (1.5%) were found in BRCA1/2 (See Supplemental Data Table S3). Among the variants detected in the coding regions, 75 (39.7%) were nonsense (stop-gain) or frameshift mutations causing protein function loss, 6 (3.2%) were detected in the canonical splice sites (splice donor and acceptor), and 91 (48.1%) were missense mutations affecting amino acid sequences (See Supplemental Data Table S3).
The details of the most frequent variants predicted to be pathogenic or likely pathogenic using NGeneAnalySys™ are listed in Table 2. The pathogenic variant c.5496_5506delGGTGACCCGAGinsA (p.Val1833Serfs) in BRCA1 was most predominantly found in eight patients, followed by c.390C>A (p.Tyr130Ter), c.5445G>A (p.Trp-15Ter), c.5470_5477delATTGGGCA (p.Ile1824Aspfs), and c.922_924delAGCInsT (pSer308fs). Five pathogenic variants in BRCA1 (c.5467+1G>A, c.5266C>T, c.4981G>T, c.3627dupA, and c.928C>T) were found at least thrice. For the BRCA2 gene, each two patients carried c.1399A>T (p.Lys467Ter), c.5576_5579delTTAA (p.Ile1859-Terfs), and c.8951C>G (p.Ser2984Ter) variants.
Table 2
Pathogenic/likely pathogenic mutations identified in multiple samples
![]()
5. Comparison with Sanger sequencing assay
The BRCAaccuTest™ results at all 199 positions were compared with those of the Sanger sequencing assay. Within the 198 individual samples, 1,640 mutations and 37,762 wild-type calls were detected. The agreement analysis results are shown in Table 3. Variant-level concordance (PPA and NPA) was 100% for all results with 95% confidence intervals of 99.8–100.0% for mutations (PPA) and 99.9–100.0% for wild-type location (NPA).
DISCUSSION
Many diagnostic laboratories use NGS technology to enhance throughput and reduce turn-around time and cost. However, NGS may introduce complexity resulting from the selection of components of the BRCA test workfiow, including the NGS platform, enrichment methods, and bioinformatic analysis processes.
The report described an NGS-based IVD reagent and analysis software for BRCA1/2 gene testing that are more efficient than the existing Sanger sequencing method. Further, this study tested the analytical performance of an NGS approach for BRCA1/2 mutation analysis in HBOC patients. The comparison of NGS and the gold standard Sanger sequencing revealed 100% sensitivity and 100% specificity in all coding exons of BRCA1/2 and 10 bp into the introns from intron/exon junctions of 212 HBOC samples.
NGS exhibits great potential by allowing rapid mutational analysis of multiple genes in HBOC [18]. However, a reliable tool is desirable to take advantage of these opportunities to extract information from the big data generated by NGS. False-positive SNVs were directly adjacent to the 3′-end of one of the first PCR primers and were detected in only one strand [19, 20]. False-negative results have also been reported, missing the pathogenic BRCA mutation owing to differences in the NGS platform and library formation [21]. Hence, caution is required while sequencing to avoid false-positive results.
The bioinformatic pipelines provide an automated workfiow for the processing of NGS data for BRCA1/2 genes using variant filters adapted to the amplicon-based target NGS data [18]. However, in the present study, the entire NGS technique from wet laboratory steps to bioinformatic analysis was managed using a single kit; this process is critical to maintaining accuracy. BRCAaccuTest™, including NGeneAnalySys™, is an all-in-one method comprising the complete NGS procedure from library preparation to automated post-processing bioinformatic pipeline and interpretation.
This study has a limitation stemming from the exclusion of large genomic rearrangement in the NGS assay for BRCA1/2. PCR enrichment is unsuitable for the reliable detection of copy number variants (CNVs) [9]. A recent study showed that the detection of BRCA1/2 rearrangement is still challenging, especially in the case of exon 2 in BRCA1 , where false-positive CNV calls may be observed [22]. The assay for BRCA1/2 rearrangements by NGS requires substantial improvement in algorithms in order to be used in clinical practice.
In conclusion, BRCAaccuTest™ can be exploited for clinical purposes, as it provided positive, negative, and overall diagnostic consistency of 100% with Sanger sequencing at a significance level of 5%. In addition, NGeneAnalySys™ can be used clinically to analyze data generated from BRCAaccuTest™ for screening pathogenic variants of the BRCA1/2 genes.
 XML Download
XML Download