

 PDF
PDF Citation
Citation Print
Print



암의 진단분류와 유전체 분석
1. 암의 진단분류는 왜 중요한가?
질병의 진단 분류는 왜 중요한가? 특정 질병을 정의하는 것 혹은특정 질병으로 환자를 진단하는 것은 해당 질환의 진행 경과에 대한 정보, 예후에 대한 정보를 알 수 있게 해주며, 적절한 치료 방법을 제공할 수 있게 해준다. 따라서, 의료의 중심체계이다[1, 2].
암의 전통적인 진단 방법은 암 조직의 생검을 통해 세포의 유래(cell-of-origin)를 판단하여 유래한 조직의 병리학적 기준에 따라 진단이 이루어진다. 그리고, 이러한 분류에 따라 임상적인 특성이나, 예후 등을 조사, 분석하고 종양 치료 지침이나 방법을 결정해왔다[3]. 종양 관련 유전자에 대한 이해가 부족했던 20세기 상황에서는 조직 병리 소견에 의한 암 진단 분류는 최선의 방법이었다. 암은 유전자 이상에 의한 질환으로 1990년대에 결론이 모아졌다[3]. 암의 유전자 분석과 분자 생물학의 발달로 인해 종양들의 유전적 특성과 암의 기전에 대한 이해가 진전됨에 따라 이를 반영한 진단과 표적치료제의 개발로 이어져서 종양 질환의 치료 성적은 극적으로 향상되고 있다. 현재 암에서 유전 분석의 역할은 점점 광범위하게 이루어지고 분석의 의미는 더 중요해질 것으로 보인다. 그러면, 암의 진단 분류는 어떻게 변화할지 예측하는 것은 단순히 학문적인 관심사가 아니다. 본 종설의 목적은 현재 진행되고 있는 이런 변화를 시간순서대로 기술하면서 현재의 이슈를 정리하고 전망하고자 한다.
2. 암의 유전 연구와 치료제
1) 만성골수성백혈병(Chronic Myeloid Leukemia, CML): 암의 진단기준과 표적치료제 등장
만성골수성백혈병은 백혈병 중에 19세기에 기술된, 가장 먼저 명명된 백혈병으로서 1960년대에 Nowell과 Hungerford는 Ph 염색체를 발견하였고, 1973년 Rowley 등에 의해 9번 염색체와 22번 염색체 사이에 전위가 된 사실을 알게 되었다. 그리고, 1984년 Eli Canaani 등에 의해 bcr-abl 융합유전자(fusion transcript)가 만성골수성백혈병 환자의 세포에서 존재하는 것을 알게 되었다. 그리고, 1990년대 초 bcr-abl 융합 유전자를 가진 유전자변형 쥐(transgenic mouse)에서 백혈병과 고형암이 생기는 경향을 발견하였다[4]. 이러한 연구 성과를 바탕으로 1990년대 초 bcr-abl kinase activity를 억제하는 화합물이 만성골수성백혈병을 치료할 수 있다는 추정을 할 수 있었고, Brian Drucker 등은 Ciba-Geigy사(현재 Novartis사)와 협력하여 bcr-abl 종양단백을 억제하는 화합물(tyrosine kinase inhibitor, TKI)을 개발하였다. 그리고, 1996년 이 화합물을 이용한 전임상을 통해 만성골수성백혈병 암세포에만 선택적으로 억제 작용이 있음을 보고하였고, 1998년부터 임상에 사용이 시작되었다[5, 6]. 이 화합물이 Imatinib이다. Imatinib는 임상에서 10년 전체 생존율(overall survival rate)이 83.3%로서[7], 이전까지 사용되었던 병합요법(interferon alpha와 cytarabine)보다 탁월한 치료 성적을 보였다[8]. 이것은 암의 유전 연구에 이어 이를 바탕으로 유전 특성에 기반한 표적 치료제 개발의 첫 사례였다. 이후에도 항암제 내성을 극복하는 2세대 TKI인 dasatinib [9], nilotinib [10] 등이 개발되어 항암제 개발과 이용에 대한 새로운 패러다임을계속 열어 나가고 있다(Fig. 1).
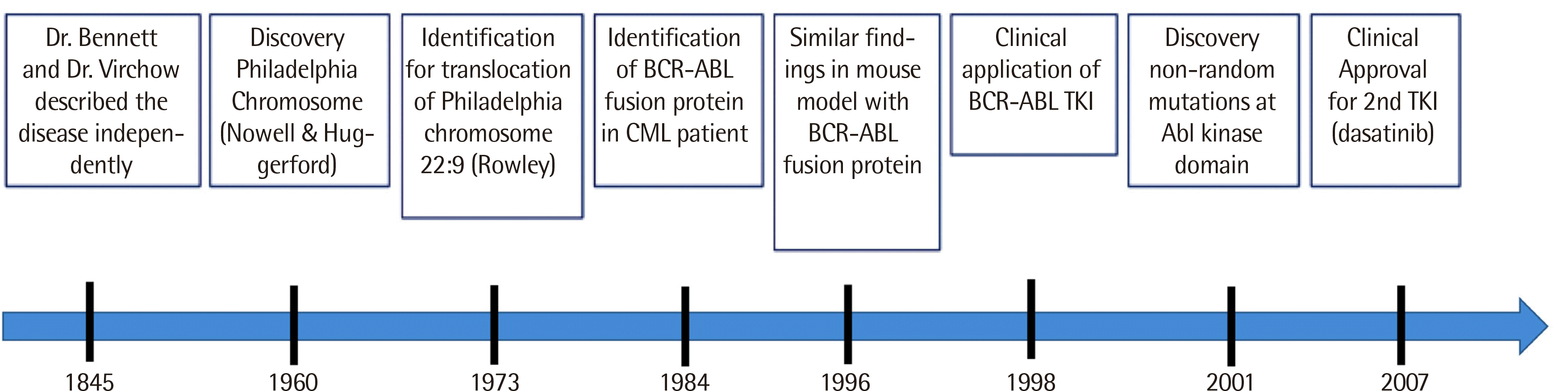 | Fig. 1Advances in understanding chronic myeloid leukemia (CML) and strategies for its treatment. CML is the first described leukemia. Dr. Bennett was the first researcher to report CML, but he had regarded it as an inflammation; Dr. Virchow suggested that it could be cancer.
Abbreviation: TKI, tyrosine kinase inhibitor.
|
만성골수성백혈병과 다른 만성골수증식성질환(myeloproliferative neoplasm, MPN)을 포함한 혈액 종양 질환을 나누는 기준은 bcr-abl 융합유전자 존재 여부이다. 이를 제외한 어떤 임상적 소견도 이를 대체할 수 없으며 bcr-abl 융합유전자의 존재를 얼마나 정밀하게, 미량까지 평가하는 것이 진단의 기준이 된다[11]. 이것은 bcr-abl 융합유전자가 존재하는 환자에게 TKI 약제를 사용하게 되고, 이 경우 탁월한 치료 성적을 기대할 수 있기 때문이다. 즉, 만성골수성백혈병의 정의와 치료제 선택은 동전의 앞뒤와 같은 밀접한 관련을 가지고 있다.
2) 유방암: 고형암의 사례
유방암은 유방에서 발생하는 모든 악성 종양을 말한다. 유방암의 치료는 1970년대까지 근치유방절제술(radical mastectomy)이 표준으로 간주되었다. 그러나, 1981년 이뤄진 무작위 임상시험(randomized clinical trial)에서, 2 cm 이내의 조기암에서 근치유방절제술과 유방보존수술(breast preserving surgery) 사이에 치료 성적의 차이가 없다는 결과가 보고되었다. 이를 기반으로 유방암의 분류 기준을 생물학적 기전에 바탕으로 한 연구가 진행되었다[12, 13]. 1995년에 국제 공동협력기구(Early Breast Cancer Trialists’ Collaborative Group)가 만들어져 1990년 이전에 시행된 55개 임상 시험군 37,000명 유방암 환자 결과를 모아서 분석한 결과에서 림프절 전이, 발병 연령, 폐경 여부와 관계없이 Tamoxifen 약제가 에스트로겐 수용체 양성(estrogen receptor positive, ER+) 유방암 환자에서 치료 효과가 있다는 것이 확인되었다[14]. 더욱이, Tamoxifen에 이어서 aromatase 억제제, CDK 4/6 억제제 등이 개발되어 치료 성적의 지속적 향상이 이루어지고 있다(Table 1) [15].
Table 1
Advances in biological knowledge and changes in treatment methods in breast cancer
![]()
병행해서 human epidermal growth factor receptor 2 (HER 2)의 발견은 이를 표적으로 하는 trastuzumab (herceptin) 약제를 개발하는 계기가 되었고, 이후 Her2/neu 유전자의 증폭이 있는 유방암 환자 중에서 trastuzumab 약제가 치료 성적의 향상을 보이는 결과를 보였다. 유방암 환자 중에서 Her2/neu 증폭을 보이는 환자는 대략 20%를 차지한다[15-18].
최근에는 BRCA1/2 유전자의 생식세포 돌연변이로 인한 유방암 환자에서 poly ADP-ribose polymerase (PARP) 억제제가 효과를 보이는 것이 확인되어, BRCA1/2 돌연변이 여부가 유방암에서 중요한 평가 기준이 되고 있다[19].
따라서, 현재 유방암의 질병 진단에서 중요한 정보는 치료제 선택에 영향을 주는 에스트로겐 등 호르몬 수용체의 발현 여부, Her2/neu 유전자 증폭 여부, BRCA1/2 유전자 변이 여부이다[15, 18]. 즉 전통적인 병리 소견 외에 이들 유전자의 이상 여부가 치료제 및 치료 방침 선택에서 중요한 판단기준을 제공한다. 이런 성과를 바탕으로 유방암에서 유전 연구는 더욱더 가속화되고 있다.
만성 골수성 백혈병과 유방암에서 유전자 이상을 반영한 치료제의 등장과 치료 효과의 향상은 자연스럽게 다른 암들의 유전체 분석을 자극하게 되어 The Cancer Genome Atlas (TCGA) 사업을 추진하게 되었다.
3. 모든 암에 대한 체계적인 유전체 분석: TCGA 사업의 수행
TCGA 사업은 2006년 미국 국립보건원(National Institutes of Health)의 후원 아래 처음 폐, 난소 및 교모세포종의 암 유전체 분석을 시작하여 성공적인 결과를 얻은 후, 2009년 암종 전체로 확대하여 33개 암종에서 11,000 증례에 대한 분석이 진행되었다[20]. 이 사업의 내용은 대단위 유전체 염기서열 분석과 전사체, 단백체등을 포함한 융합 데이터를 생산하여, 암을 초래하는 변이들의 데이터를 모으고, 분석하고자 하는 공공 사업이다. 목표는 이와 같은 데이터를 모은 결과로 진단방법과 치료방법의 향상과 암의 예방을 목적으로 하였다[21]. 이 사업에서는 mRNA sequencing, microRNA sequencing, array-based DNA methylation sequencing, reverse-phase protein array (RPPA), structural copy-number alterations (SCNA), somatic mutations 등 6가지 방법에 기반한 데이터들이 생산되어, 이들 데이터로부터 현재까지 35편 이상 개별 암종의 유전체 분석 논문이 발표되었다[22]. 그리고, 광범위 암 분석 사업(Pan Cancer Analysis project)을 2012년 시작하여 11,000개 암종의 분석결과를 2018년 발표하였다[20].
4. TCGA 사업과 암의 진단 분류
암의 유전체 지식을 이용한 암의 진단방법 향상은 처음부터TCGA 사업의 주요 목표였다. 처음 TCGA 사업은 암의 유전체 분석을 통해, 개별 암종에서 유전체 양상이 다른 몇 개의 아형이 존재함을 밝혔다. 그리고, 여러 암종을 모두 모아 한꺼번에 암 유전체 분석을 시도하여 기존 암 병리 분류체계와 비교하게 되었다. 현재 병리 암진단은 유래 세포(cells-of-origin)를 기준으로 하여 개별 유래 세포가 속한 조직의 병리학적 기준에 따라 개별 암의 진단 기준이 수립되어 진단이 이루어진다. Hoadley 등[23]은 TCGA에서 생산된 데이터를 기반으로 12개 암종에서 3,527검체의 종양을 분류하였다. 유전체 분석에서 사용한 플랫폼(platform)은 mRNA sequencing, microRNA sequencing, array-based DNA methylation sequencing, reverse-phase protein array (RPPA), structural copy-number alterations (SCNA), somatic mutations의 여섯 가지를 이용했다. 이들 중 체세포 돌연변이(somatic mutation)를 제외한 5개 플랫폼에서 개별적으로 무리(cluster)를 지은 후, 이들의 무리를 다시 2차적으로 무리를 지은 후 group을 분류하였다. 이들은 이 방법을 COCA (clustering of cluster algorithm)로 명명하였다. 이 결과 분자유전학적 분류가 전통적인 병리학적 분류와 약 10% 정도에서 차이가 있었으며, 이는 분자유전학적 분류가 질병의 임상적 특징을 보다 잘반영할 수 있다는 점을 시사한다. TCGA가 더 많은 검체에 대한 데이터를 생산함에 따라 2018년 33개 암종의 10,522개 검체를 대상으로 최소 4개의 플랫폼 즉, 염색체 이수성(aneuploidy), DNA 메틸화(DNA methylation), mRNA와 miRNA expression과 일부 검체(7,858/10,522)에서 RPPA 단백 데이터를 생산하여 더 개선된 방법으로 분류를 시도하였다(Fig. 2) [24]. 이들은 5개 플랫폼을 이용하면서 체세포 돌연변이 정보는 분류에 반영하지 않고 개선된 COCA 분류 방법을 통해 33개 암종을 28개 군으로 분류를 하였다. 이들분류를 통해서 드러난 특징은 암종의 유래 세포가 중요하기는 하나, 이것이 종양의 분류에 전적인 요인은 아니라는 것이다[24]. 예를 들면 같은 장기에서 유래한 암이라고 하더라도 메틸화 양상은암의 조직학적특이 양상을 보이는 사례도 있으며, 다른 장기에서유래한 암이라도 같은 메틸화양상을 보이고 있는 경우도 있다. 28개 군 분류에 대해 복제수 변이는 전체 분류에서 47%, 전사체 분류(mRNA와 miRNA)가 42%, DNA 메틸화가 11%의 기여도를 보였다[24]. 이들을 통해 알게 된 사실은 유두상 신세포암(papillary renal cell carcinoma)과 투명세포 신세포암(clear renal cell carcinoma) 하나로 묶이는 Pan-Kidney 군으로 분류가 되며[25], 결장의 선암(colon adenocarcinoma), 직장의 선암(rectum adenocarcinoma), 위의 선암(stomach adenocarcinoma), 식도의 선암(esophageal adenocarcinoma)을 포함하는 소화기 암들은 miRNA, mRNA, RPPA 양상을 공유하나, DNA 메틸화 양상은 암종들 사이에 특징적으로 차이가 난다[26]. 폐의 편평세포암(lung squamous cell carcinoma), 두경부의 편평세포암(head and neck squamous cell carcinoma), 자궁경부의 편평세포암 및 자궁경부내막의 선암(cervical squamous cell carcinoma and endocervical adenocarcinoma), 식도암(esophageal carcinoma), 방광 및 요로상피암(bladder urothelial carcinoma)을 포함하는 편평세포군 암(Squamous histology cancer)은 miRNA, mRNA, RPPA 양상은 유사하나, 염색체 이수성과 DNA 메틸화 양상이 서로 다르다[27]. 침윤성 유방암(invasive breast cancer)과 난소 장액낭선암종(high-grade ovarian serous cystadenocarcinoma), 자궁내막암(uterine corpus endometrial carcinoma, UCEC), 자궁 편평세포암(cervical squamous cell carcinoma, endocervical adenocarcinoma, CESC), 자궁암육종(uterine carcinosar-coma, UCS)을 포함하는 Pan-Gynecologic 암종군에서는 단백체 수준에서는 유사하나, miRNA, mRNA, DNA 메틸화양상은 장기에 따라 다르다[28]. 암 종의 염색체 이수성은 전체 암종 검체의 88%에서 나타난다. 갑상선 암과 급성골수성백혈병은 염색체 이수성이 낮고, 교모세포종(astroblastoma), 자궁암육종, 고환 생식 세포종양(spermocytoma)에서는 염색체 이수성이 높았다. 그리고, 이수성과 점 돌연변이의 수와는 반비례 관계를 보였으나, 이는 high microsatellite instability (MSI-H)에 대한 대장암, 자궁내막의 효과이고, 이러한 과돌연변이(hypermutation) 종양을 제외하면, 이수성과 점 돌연변이는 비례관계를 보였다. 이수성이 높은 종양은 백혈구 침윤(leukocyte infiltrate)이 낮아 면역신호 세포들의 발현과는반비례 관계를 보였다[29]. 또 편평세포군에서는 염색체 3p 소실이흔히 발견되었다[29]. 최근 면역 치료가 중요한 종양 치료 약제로등장함에 따라 면역측면에서 암종을 분류하면, 상처 회복(wound healing), 인터페론 감마 우위형(IFN-γ dominant), 염증형(inflammatory), 림프구 고갈형(lymphocyte depleted), 면역 침묵형(immunologically quiet) 및 전환성장인자 베타형(TGF-β dominant)등 6개 유형으로 나누어졌다[30]. COCA 법을 이용한 분류는 일단유래세포가 반영하는 정보가 압도적이라고 할 수 있으나, 그 외 정보를 더 제공함으로써, COCA 법을 이용한 유전체 분류방법이 예후 등의 임상적 변수에서 더 우수한 성과를 보인다고 보고하고 있다[23]. COCA 분류 방법은 특정 돌연변이 여부를 반영하기보다는활성 혹은 저하된 대사 경로 중심으로 분류한다. 또한 이들 분류를 좀 더 실용적인 접근 방법을 통해, 예후 혹은 치료 후 성적 기반으로 분류하는 방법이 제안되었다[31].
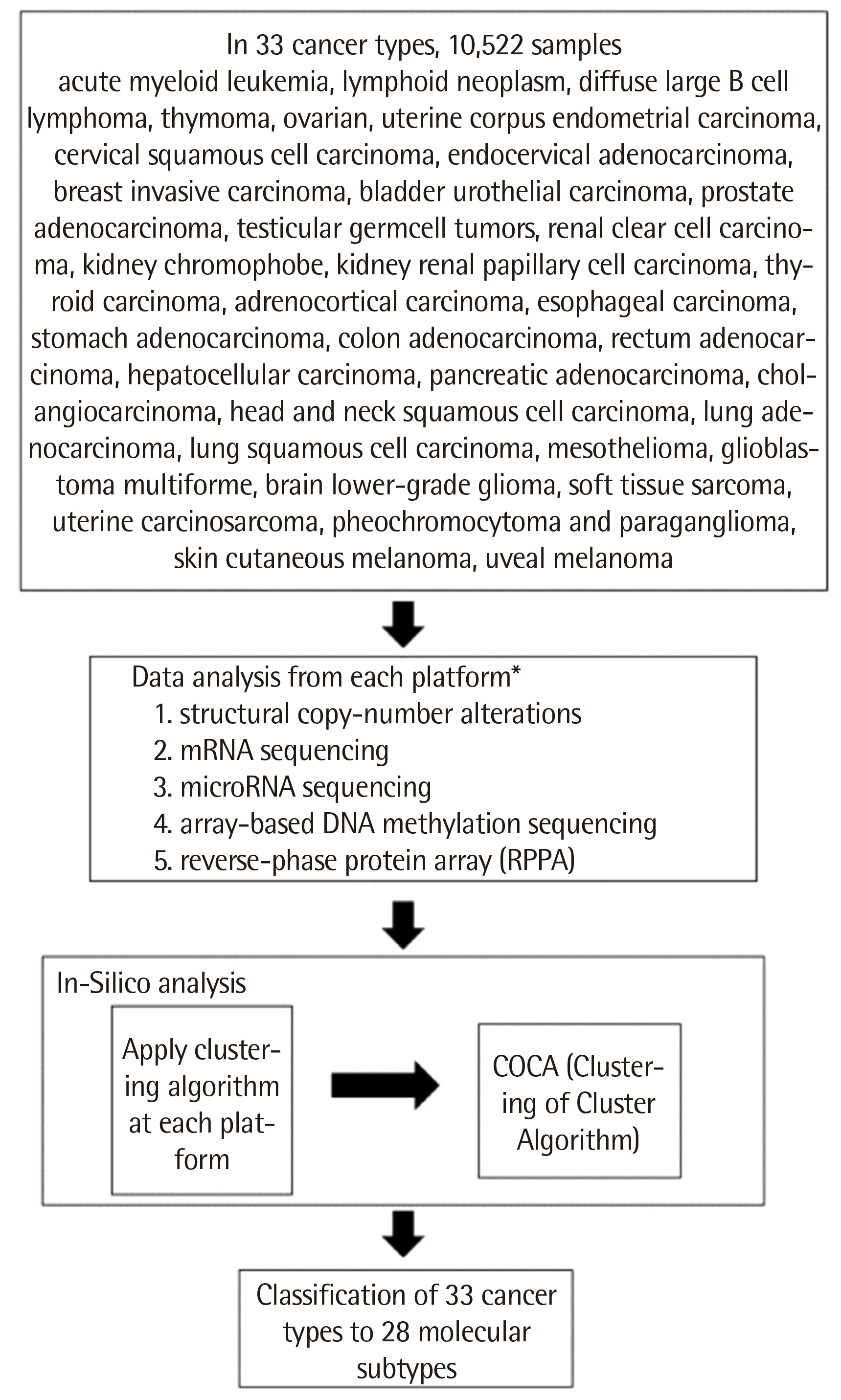 | Fig. 2The progress of cancer classification of TCGA program. *Somatic mutations were included when 12 cancer types were classified using other platforms (besides the 5 platforms) but were excluded when 33 cancer types were classified. Moreover, because the RPPA platform was used for a few specimens (7,858/10,522), only four platforms were used to analyze all specimens.
|
5. 돌연변이, 암의 분류 문제
특정 돌연변이의 존재는 표적치료제의 대상이 되고, 이들 표적치료제는 암 치료에서 획기적인 성과를 가져왔다. 그러므로 현재 시점에서 특정 돌연변이의 유무는 실질적인 암의 진단에서 중요한 위치를 차지한다. 2019년 말 기준으로 15개 암종에서 80개의 표적을 대상으로 165개 치료제가 임상시험이 진행되고 있다[32]. TCGA에서 10,000개 암 검체 분석 결과를 보면, 표적 치료로서 인정이 되는 표적의 돌연변이를 가지고 있는 암 검체는 전체 암의 약 50%를 차지하며 두 개 이상의 표적의 돌연변이를 가지고 있는 암 검체도 약 30%를 차지한다[33]. 표적치료제의 개발이나 임상 도입이 최근에 시작된 것이라는 점을 감안하면, 모든 암 종에서 선택할 수 있는 다양한 표적치료제가 준비되는 날이 가까운 미래에 있을 것으로 낙관적인 예상을 할 수 있다.
미국 식약처(FDA)는 2017년 처음으로 암 유래조직에 대한 고려 없이 생체 표지자(biomarker)의 존재만으로 치료를 하는 조직 불문 약물(tissue-agnostic drug) 개념의 약제 사용을 승인하였다. MSI-H 혹은 mismatch repair (MMR) deficient 암에 대해 승인을 받은 pembrolizumab (keytruda)가 바로 그 사례이다[34]. 이후 NTRK 융합단백질을 표적으로 하는 larotrectinib [35], ROS1 양성비소세포암과 NTRK 융합단백질 양성암을 표적으로 하는 entrectinib [36] 등이 승인을 받았다. 그렇지만 이미 BRAF V600E 억제제는, BRAF V600E 변이 양성인 흑색종에서는 표적치료제로서 역할을 하나, BRAF V600E 양성인 대장암에서는 효과가 없는 것을 발견하였다. 대장암에서 BRAF를 억제하게 되면, EGFR 신호 경로가 활성화되어 암세포 증식이 촉진된다. 실제로 흑색종에서는 EGFR 신호 경로의 발현이 낮으므로 BRAF 표적 치료제가 암 억제 역할에 효과적이다[36, 37].
또한 돌연변이에 대한 표적 치료제를 사용하게 되면, 내성이 발생한다. 내성의 기전은 해당 표적 유전자의 돌연변이로 표적치료제 접근을 억제하거나, 다른 유전자(들)의 돌연변이로 발생하게 된다. 그러므로 단순 돌연변이의 존재 여부로 암의 분류체계를 시도하는 것은 한계가 있다. 2021년 미국 식약처에서 비소세포 폐암 환자 중 KRAS G12C에서 사용이 허가된 sotorasib의 경우[38], 이전임상 시험 결과[39]에서, KRAS G12C 돌연 변이를 가진 환자들 중비소세포폐암에서는 반응이 좋으나, 상대적으로 대장암에서는 치료 반응이 저조하다는 보고를 보면, BRAF 억제제의 사례가 예외적인 사례가 아닐 가능성이 높다는 것을 시사한다.
6. 암의 분류의 전망
암은 유전자 이상에 의한 질환이다. 따라서, 유전자 이상의 종류에 따라 암종의 분류가 되는 것은 당연하다고 할 수 있다. 1850년에 Virchow가 세포 병리를 제안하고, 세포 및 조직 병리 기준이 암의 진단과 분류 기준이 된 이래, 암에 대한 이해는 많은 발전이 이루어졌다. 이제 암에 대한 유전적 이해가 깊어지고, 표적치료제가 등장함에 따라 암의 유전학적 분류 체계를 수립하는 것은 다음 단계의 발전을 위해서 필요한 일이다. 현재는 대개 하나의 표적치료제를 중심으로 항암 치료를 시행하고 있으나, 표적치료제가 계속 개발되고 있으므로 치료 표적이 될 수 있는 변이가 점점 더 많아지고, 이들 표적 가운데, 어떤 표적들에 대해 어떻게 복합적으로 표적치료제를 사용하는 것이 좋을지 이슈가 될 것이다. 따라서, 암의 유전적 이상에 따른 암종의 진행 기전의 차이를 이해하고, 이에 따른 정밀 의료를 구현하기 위해서는 개별 암에서 활성화된 경로에 대한 이해와 체계가 전제 조건이 될 것이다.
Go to : 
 XML Download
XML Download