

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Multiple myeloma (MM) is characterized by the accumulation of abnormal clonal plasma cells (PCs) in the bone marrow (BM), which results in BM failure. Treatment includes chemotherapeutic drugs and autologous stem cell transplantation (ASCT), and due to significant advancements in the last decade, most patients achieve a complete response (CR) and high overall survival (OS) rates. However, MM remains an incurable disease, and most patients relapse within five years [1, 2]. Standard monitoring assays such as serum and urine paraprotein (M-protein) detection in the peripheral blood (PB) and the percentage of PCs in the BM are used to define CR [3, 4]. However, these methods are limited in their sensitivity [5, 6] and newer methods have been developed to detect and characterize abnormal PCs including multiparametric flow cytometry analysis (EUROFLOW) (MFC) and polymerase chain reaction (PCR)/next generation sequencing (ASO-PCR and NGS) to detect clonal B cell receptor rearrangements of specific abnormal PC clones [7]. These methods have detection limits of 0.01–0.001% of PC populations and have indicated that minimal residual disease (MRD) is present (defined as abnormal PCs) in the BM of patients with CR, and that this MRD is responsible for clinical relapses [1, 6, 8-11].
These newer methods have their limitations, as they require patient-specific adaptations and BM aspirate samples; additionally, they only assess the PC burden [7], and thus do not reflect the levels of the total malignant cell population that includes a stem cell component [12-16]. Circulating malignant cells lead to extramedullary disease; thus, monitoring their levels and gene expression profiles may provide a novel tool for assessing relapse and disease progression risk during therapy. A simplified quantitative reverse-transcription PCR (qRT-PCR) approach using PB samples that is applicable in all MM patients may help overcome the limitations of the current methods.
MM is a heterogeneous disease and a common molecular signature has not been established yet; however, we and others have shown a distinct relationship between the number and type of cancer/testis antigens (CTAs) expressed with advancing disease, and suggested a cascade-like expression pattern [17-20]. Additionally, the CTA “melanoma associated antigen C1” (MAGEC1) was identified as a diagnostic marker of symptomatic MM (SMM); it was the most frequently expressed CTA in MM, and was found to be expressed in both early and advanced disease [20, 21]. A pro-B/pre-B stem cell progenitor has been reported to express this protein, implicating MAGEC1 in carcinogenesis; thus, these early stem cells circulating in the PB may be the malignant progenitor cells that function as the feeder population in MM, in addition to proliferating PC populations that are detected in some patients [15]. MACEC1 expression reflects treatment responses and changes in serum M protein and serum beta-2-microglobulin (Sb2M) levels. Thus, qRT-PCR or flow cytometry monitoring of MACEC1 is relevant in chemotherapy patients, and significantly high sensitivity to low levels of disease was observed with this approach [21].
Our previous studies demonstrated that the CTA “preferentially expressed antigen of melanoma” (PRAME) was expressed in early stage disease (from stage 1), MAGEA3 in advanced disease (stages 2/3), and BAGE2 exclusively in stage 3 disease [20]. Whether the expression of these genes occurs in different clones or represents clonal evolution has not yet been established. Therefore, we wanted to investigate whether the simultaneous monitoring of multiple CTA transcripts would be more informative than monitoring MAGEC1 alone, especially regarding clonal development and relapse prediction. Thus, the primary aim of this pilot study was to characterize CTA (MAGEA3/ABL; PRAME/ABL, and BAGE2/ABL) expression profiles using multiplexed qRT-PCR in the PB of MM patients during chemotherapy, and to compare these data to the MAGEC1 transcript levels already established in the same patient cohort in our previous study [21]. Additionally, we aimed to investigate the clonality of the MAGEA3/PRAME/MAGEC1-expressing cell populations using flow cytometry to provide evidence of a cascade-like expression pattern occurring in stem cells during the course of the disease.
MATERIALS AND METHODS
MM patient samples
Patient samples were collected and RNA was processed as part of a previous CTA study [21]. Ethics approval for the analysis of these samples was obtained from the Human Ethics Research Committee at the University of Cape Town, Faculty of Health Sciences (HREC REF: 194/2012). Briefly, PB samples (10 mL EDTA) were collected every three months from diagnosis for up to two years during treatment with basic chemotherapy regimens (Table 1). The PB mononuclear cells (PBMCs) were isolated using density centrifugation with Ficoll Histopaque (Sigma-Aldrich, St. Louis, MO, USA) and analyzed via flow cytometry to determine the MAGEC1- positive populations as previously described [21]. The RNA was extracted using the QIAamp RNA blood mini kit (Qiagen, Hilden, Germany), treated with the TURBO DNase-free Kit (Ambion, Waltham, MA, USA) to remove residual DNA, and checked for purity and integrity (quality: 260/280 nm ratios >1.8; RNA integrity number (RIN) values: 7–10 [22], MIQE guideline compliant [23]) as per a previous study [15]. The stored RNA from 10 patients was used in the present study. Serum M protein and Sb2M levels were measured by the National Health Laboratory Service (NHLS)/GSH (South Africa) using accredited methodology. We compared our novel monitoring method with standardized hematological monitoring methods. Notably, the monitoring of serum free light chains was not available at our state hospitals during the study period.
qRT-PCR multiplex assays
Multiplex assays were developed and validated as per MIQE guidelines, including the assessment of amplification efficiency and specificity, and limits of detection. Reverse transcriptase (RT) reactions using 1.5 mg RNA and random hexamers were performed as previously described [21]. The primers for coamplifying MAGEA3/ABL, PRAME/ABL, and BAGE2/ABL are listed in Table 2. PCR reactions (25 mL) contained 0.8 mM dNTPs, clear GoTaq Flexi buffer, 1.5 U GoTaq Hotstart (Promega, Madison, WI, USA), 4 mL cDNA (20% of RT reaction), 4.5 mM or 2 mM (MAGEA3) MgCl2, 0.4 mM of each CTA primer/0.2 mM CTA probe and 0.2 mM of each ABL primer/0.1 mM ABL probe. The cycling conditions were as follows: 95°C for 5 min, 40 cycles of 95°C/30 s, 60°C/40 s (57°C for BAGE2), and extension at 72°C/40 s (MAGEC1/ABL amplification was performed as per a previous study) [21]. All reactions were performed on the Rotor-Gene 6000 PCR machine (Qiagen) and analyzed using the Rotor-Gene 6000 series software 1.7. Cq values were determined using appropriate thresholds depending on the probe fluorescence. Target genes were quantified relative to ABL (reference gene) expression (normalized ratio) using a modified Livak method [24], as follows: Ratio (reference/target)=2Cq(reference)-Cq(target) or ΔCq. All RNA samples were analyzed in duplicate (acceptable CV, <20%). The assays were able to detect >5 copies of each CTA transcript type; positive/negative (pos/neg) Cq cutoff values were based on assay-specific controls. Cq values generated from logarithmic curves were scored as positive. An ABL control indicative of 10,000 ABL copies/reaction was included in each experimental run; samples with ABL Cq<ABL Cq of controls (indicating potential RNA degradation) were excluded from further analysis.
Flow cytometry analysis of PBMCs
Approximately 2×106 ethanol-fixed PBMCs from two patients prepared during the previous studies [15, 21] were washed twice in 40 mL ice-cold 1× PBS/1% fetal calf serum (FCS), collected via centrifugation (1,500 rpm for 5 min), and re-suspended in 500 mL cold 1× PBS/0.5% bovine serum albumin (BSA) buffer. Approximately 100 mL of this cell suspension was incubated with the following antibody combinations for 30 min at 4°C in the dark: MAGEC1- Alexa488/CD34-PE/CD45-PerCP and MAGEA3-Alexa647 or PRAME-Alexa647 and MAGEA3-Alexa647/PRAME-Alexa488/ CD34-PE/CD45-PerCP; MAGEC1-Alexa488/CD138-PE/CD45PerCP and MAGEA3-Alexa647 or PRAME-Alexa647. All antibodies were used at a final concentration of 5 mg/mL. MAGEC1 (Abcam, clone ab115351), MAGEA3 (Abcam, clone ab38496) and PRAME (Abcam, clone ab135600) antibodies were fluorescently labeled with the Mix-n-Stain Alexa 647 kit (Sigma-Aldrich). The labeled cells were washed with 1 mL cold 1× PBS/0.5% BSA buffer at 1,500 rpm for 5 min; the pellet was re-suspended in 1 mL cold 1× PBS/0.5% BSA buffer and stored in the dark until analysis. The cells were analyzed on a BD FACSCalibur flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA) using the Cell Quest Pro software, applying parameters and gates that had been previously optimized to minimize spectral overlap and increase sensitivity [15, 21].
RESULTS
Detection of MAGEA3, PRAME, and BAGE2 transcripts
PRAME transcripts were detected in all the 10 patients at diagnosis, and were observed in 81% of all the RNA samples. This demonstrated the high prevalence of expression of this CTA and a possible role in early pathogenesis, in agreement with our previous findings [20]. Similarly, all patients showed MAGEC1 expression at diagnosis, and 85% of the RNA samples showed detectable transcripts [21]. In contrast, only five patients showed MAGEA3 expression at any time point (positivity rate, 20%). However, this was not unexpected, as MAGEC1 and PRAME expression has been linked to early stage disease [17, 18, 25], and MAGEA3 with more advanced disease [26, 27]. BAGE2 has been reported in advanced (stage 3) disease only [20]; in line with the above, we observed BAGE2 in three patients with clinically progressing disease.
PRAME and MAGEA3 expression levels varied widely, demonstrating positive normalized ratios over a 4-log range (0.00001–0.87 and 0.0005–4.31, respectively); these data were comparable to MAGEC1 data [21]. However, BAGE2 expression was much more limited, with positive results expressed in the 2-log range (0.0008–0.0206).
Correlation of PRAME expression with clinical parameters and MAGEC1
To investigate whether monitoring PRAME transcripts could provide clinically relevant information, the expression pattern of PRAME in each patient was correlated with biochemical parameters and MAGEC1 qRT-PCR results.
PRAME expression was detected in all patient samples where serum M protein and/or Sb2M levels were above the clinical threshold [28] (indicating active disease), or had stabilized or returned to baseline levels following therapy (patient achieved CR); thus this new approach has higher sensitivity for detecting residual disease than the techniques currently employed at our facilities. Significant changes in the levels of these protein markers were at least mirrored, but more importantly often preceded by changes in PRAME expression (examples shown in Figs. 1, 2). The high frequency of detection of PRAME made it an attractive target to assess circulating disease, especially where patients appeared clinically normal following therapy, but later progressed.
An example of this is shown in Fig. 1 (MM4), where the patient had stage1 disease at therapy initiation; the serum M levels were always within the normal range (<8.9 g/dL) and the Sb2M levels decreased to clinically normal levels (<1.8 g/dL) after therapy. While PRAME levels dramatically decreased initially, it was still detectable throughout therapy (while no clinical signs of disease were recorded); a large 2-log increase was observed six months prior to a 10-fold increase in paraprotein and the detection of further lytic lesions (at follow-up).
Even in cases with stable Sb2M and serum M levels following therapy for a significant period of time (Fig. 1 MM6), monitoring the patient with qRT-PCR revealed that their condition deteriorated before clinical changes were observed; a 2.6-fold increase in PRAME transcript levels was observed at least 10 months prior to the development of significant anemia (indicative of BM failure) and renal damage. The patient died soon after the last time point, demonstrating that monitoring of the Sb2M/serum M levels alone failed to predict the clinical complications early enough to avoid adverse outcomes.
PRAME and MAGEC1 expression followed similar patterns in this patient cohort; PRAME levels decreased simultaneously with MACE1 in response to therapy, and increased at the same time (Fig. 1 MM1/MM6, Fig. 2 MM10) or shortly after MAGEC1 (Fig. 1 MM4, Fig. 2 MM3/MM8). Monitoring PRAME levels in conjunction with MAGEC1 levels did not offer an earlier relapse prediction compared to monitoring MAGEC1 alone. However, the increases in PRAME levels were sometimes more pronounced than those in MAGEC1 levels, and served to confirm MAGEC1 data and disease progression. In line with the hypothesized occurrence of a CTA cascade with disease progression, PRAME expression may indicate early clonal changes that may influence malignant cell behavior.
Correlation of MAGEA3 expression with clinical parameters and MAGE C1/PRAME expression
Although the frequency of MAGEA3 detection in this MM patient cohort was only 20%, any detectable expression was linked to the development of anemia (Hb levels <12/13 g/dL), with an average Hb level of 10.6 g/dL, as previously observed [20]. Its expression was also strongly linked to clinical progression of the disease (plasmacytoma, severe anemia, and death) (see Figs. 2, 3). Notably, in patient MM 8, MAGEA3 expression provided valuable information about disease states and imminent disease progression, which was not apparent with standard biochemical monitoring. At diagnosis, the biochemical parameters were significantly deranged, and the patient was diagnosed with stage3 (ISS) disease; elevated renal failure markers (creatinine), anemia, and bone lesions (RAB) were also observed. As per a previous report linking reduced Hb levels with MAGEA3 expression [20], this CTA was detected at diagnosis (Hb level at diagnosis, 6 g/dL). Following chemotherapy, the patient appeared to improve clinically (serum M and anemia). However, MAGEA3 remained detectable and transcript levels increased significantly during the last three months of the patient’s disease leading up to the detection of a plasmacytoma (extramedullary disease), and the subsequently died.
The relative expression of MAGEA3 was also compared to that of MAGEC1 and PRAME in the relevant patients (MM 3, 4, 8, 9, and 10); while the increases in both MAGEA3 and MAGEC1 transcript levels correlated with imminent death or clinical relapse, the increase in MAGEC1 was detected earlier, and there were no time points at which MAGEA3 was expressed in the absence of MAGEC1. MAGEA3 expression increased at the same time (Fig. 2 MM3 and MM8) or after PRAME expression increased (Fig. 2 MM10, Fig. 3 MM9). Although the sample size was small, overall MAGEA3 expression data offered no additional advantage over MAGEC1 data for cell-level monitoring, but could be used to detect clonal changes that may affect therapy responses and possible spread of disease.
Correlation of BAGE2 expression with MAGEC1 and clinical parameters
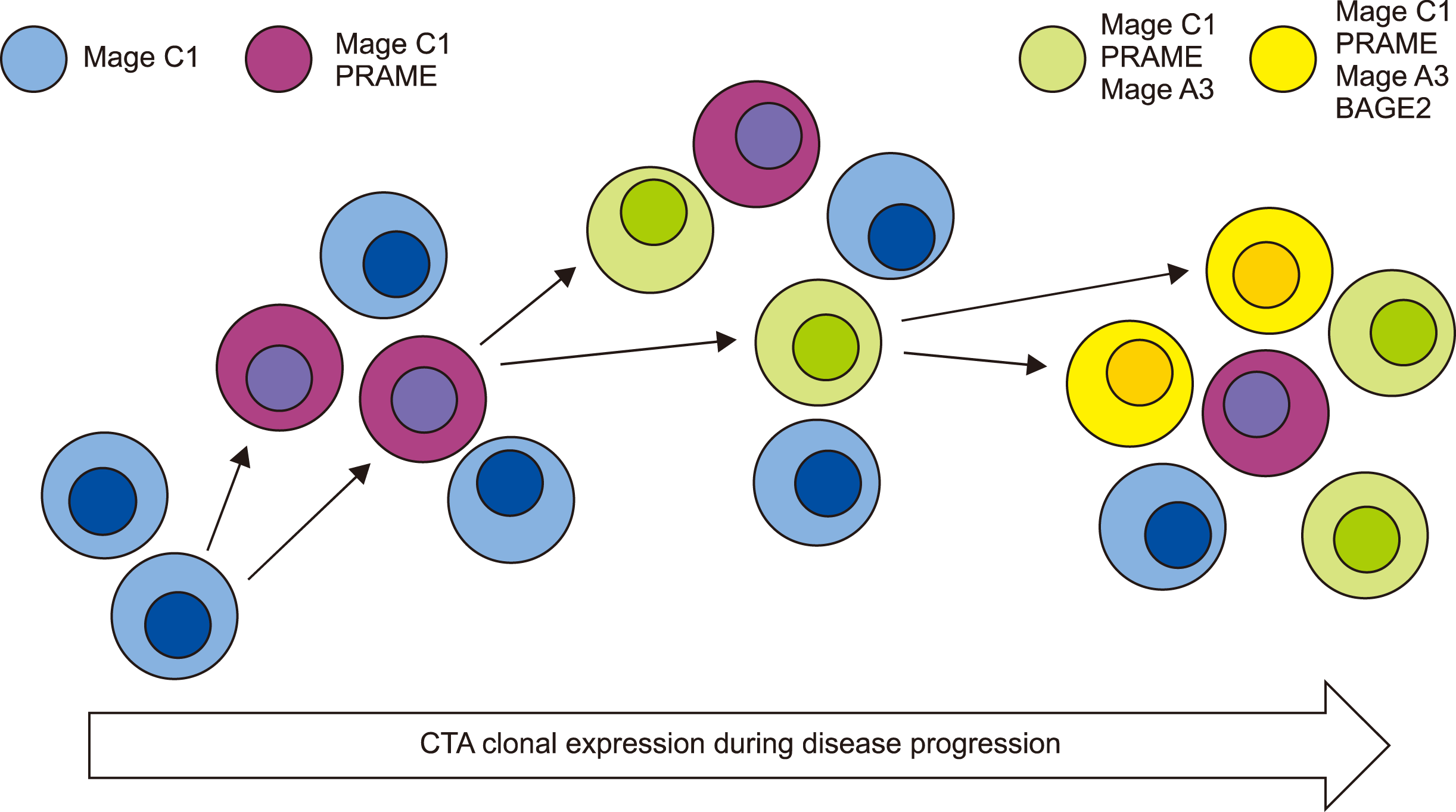
Although BAGE2 expression in our patient cohort was extremely limited, its expression was clearly associated with known indicators of stage 3 disease (as per the DSS and ISS systems), namely elevated creatinine levels (>90 mmol/L), severe anemia (<10 g/dL), and/or very high Sb2M levels (>5.5 mg/L) [29, 30]. Fig. 3 shows the changes in BAGE2 expression in patients where it was detected (MM6, 8, and 9). Although detection of BAGE2 transcripts was always associated with a prior increase in MAGEC1 transcripts, its detection was associated specifically with advanced clinical disease indicators such as elevated creatinine (indicating organ damage), rather than just elevated PC levels (increased paraprotein). This could possibly indicate advanced clonal changes to the malignant cells in a cascade-like manner, which could potentially affect the proliferation rate, resistance to apoptosis, and thus overall drug responsiveness; these features can guide treatment for the predicted relapse. The CTA expression patterns in patient MM9 (Fig. 3) were in line with this cascade theory, and additional CTAs (MAGEC1-PRAME-MAGEA3-BAGE2) were expressed within the PBMC component as anemia, renal damage, and M protein levels increased.
CTA co-expression in CD34+ cells
The co-expression of multiple CTAs in this patient group indicated a cascade-like pattern as disease progressed, as previously inferred [17-20]. However, as qRT-PCR was performed on enriched PBMCs and not isolated cell populations, it was not possible to establish whether cells were undergoing sequential clonal changes, or whether different clones, or even different cell types, expressed single CTAs. Using limited residual ethanol-fixed PBMCs from two patients from the initial studies (MM3 and MM4) [15, 21] where MAGEC1-expressing cells have been extensively characterized, we additionally analyzed the expression of PRAME and MAGEA3 antigens in the matched samples (MM3 and MM4); co-expression of MAGEC1/PRAME/MAGEA3 mRNA was observed in these samples. While this study offers only preliminary results due to the limited sample pool and cell numbers, the results indicated that PRAME and MAGEA3 were only associated with the populations previously shown to express MAGEC1, namely a pro-B/pre-B progenitor population (CD34/CD19) and a PC sub-fraction (CD138+) (Table 1) [15]. We detected the co-expression of PRAME/MAGEC1 and MAGEC1/MAGEA3 in the CD34+ population and PRAME/MAGEC1 in the PC fraction (previously shown to be a proliferating population) [15]. No cells were found to express PRAME or MAGEA3 only; only small numbers of MAGEA3-positive stem cells were detected, and they were all found to be PRAME-positive, implying sequential antigen expression as opposed to activation in separate clones, although larger cell populations are required to confirm these findings.
DISCUSSION
The ability to predict and prevent relapse in MM is hindered by insufficient knowledge about the role of circulating malignant stem cells in extramedullary disease and disease progression, and clonal evolution leading to treatment resistance. Despite recent advancements, MRD monitoring techniques have limitations; affordable, non-invasive novel molecular tools that can monitor circulating disease, and can be easily applied to all patients in a standard clinic/hospital environment are needed. BM sampling for monitoring residual disease is limited in the following ways: residual disease is non-uniformly distributed in the BM, and thus, MRD can easily be missed based on the sampling site; circulating malignant cells that can lead to extramedullary disease cannot be detected. The CTAs MAGEC1, PRAME, MAGEA3, and BAGE2 are commonly expressed in MM and their expression has been correlated with different disease stages. We aimed to evaluate the potential value of each of these CTAs in monitoring circulating malignant cells, and possibly predict relapse or clonal evolution of the disease at an earlier time than standard biochemical monitoring. Serum FLC analysis was not performed on this particular patient cohort, as it has only recently been introduced in the state. The availability of NGS and ASO-PCR is limited in South African, which further stresses the need for an alternative methodology that is more widely available. The approach we use here is novel, though controversial, as the PC mass was not assessed; instead, we assessed the small population of potential malignant cell progenitors circulating in the PB (1–2%); we thus reveal a potential alternate monitoring target.
We included patients who were not eligible for ASCT and were treated with limited chemotherapy regimens. A significant proportion of South African patients with MM in the state health sector did not undergo ASCT because of advanced age and disease at presentation (>65 yr and stage 3 disease), and comorbidities (heart disease, diabetes, and liver damage) which limit the use of myeloablative induction therapies. While most patients achieved a CR during treatment, all relapsed with extramedullary disease, and additional therapy was administered. Due to lack of novel agents for MM treatment in South African state hospitals, patients were treated primarily with cycles of cyclophosphamide and dexamethasone. Relapse in these cases was thus not unexpected. In this scenario, we aimed to assess whether qRT-PCR analysis of CTA expression could help in predicting relapse at an earlier time, allowing earlier intervention. Our previous study [21] showed that MAGEC1 could be effectively used as a monitoring tool in ASCT and chemotherapy-only patients, but as CTA expression appears to occur in a cascade-like manner with advancing disease [17-20], we wanted to investigate whether there was any clinical advantage to monitoring the expression of multiple CTAs simultaneously.
PRAME/MAGEC1 co-analysis showed promising results. We observed that all 10 patients expressed PRAME at some point during their disease, in agreement with numerous previous studies [20, 25]. qRT-PCR data showed that PRAME was always co-expressed with MAGEC1, and its expression levels correlated with changes in both biochemical parameters and MAGEC1 expression. Overall, we found that MAGEC1/PRAME co-analysis provided a comprehensive picture of the circulating MM progenitor cells; fluctuations in the expression of these CTAs reflected clinical outcomes and frequently pre-empted changes in biochemical parameters. The expression levels increased prior to the detection of extramedullary disease (plasmacytomas) and even death, despite maintenance of CR. Preliminary flow cytometry analysis showed that PRAME is expressed in a sub-clone of MAGEC1 cells. However, its expression occurs in too many patient samples and potentially too early on in the disease to be a real indicator of significant clonal changes.
Detecting potential disease-related changes earlier than at the symptomatic level may be important in predicting response to salvage therapy at the time of relapse. Therefore, we investigated the expression of MAGEA3 and BAGE2; these CTAs are potentially expressed further down the hypothesized CTA cascade [20], and have been implicated in more advanced disease stages. MAGEA3 has been associated specifically with severe anemia [20] and stage 2/3 disease. The analysis of our patient group reflected this expression pattern, and the transcript expression was detected following (or concurrently with) increases in PRAME and MAGEC1. Preliminary flow cytometry data also suggested the presence of a subpopulation of PRAME/MAGEC1+ cells expressing this third CTA, although sequential analysis of patient samples was not possible. MAGEA3 expression patterns appear to indicate clonal changes in the circulating malignant cell population (70–97% CD34/CD19) which may be linked to relapse mechanisms and potentially affect responses to therapy. BAGE2, which has previously been linked to stage 3 disease only, was detected in three patients with clinical symptoms of disease progression, and therefore can also be potentially predictive of clonal evolution at a level further along than that of cells expressing MAGEA3. The study population was too small to compare between the CTA markers, and this needs to be studied further.
Clonal evolution of the “Myeloma cells” in MM is thought to be responsible for relapsed and treatment-refractory disease [16, 31, 32]. CTAs may be involved in this clonal evolution, leading to the development of sub-clones with higher proliferative capacity, more resistance to apoptotic signals, and refractory to inflammatory control. Although the function of many CTAs is unclear at present, members of the MAGE family are known to be involved in stimulating cell cycle progression (via cyclin activity), reduction of tumor suppression activity (P53 activity), and p21 activity via increased ubiquitination [7, 33-35]. Many CTAs are reported to be involved in overcoming cell cycle blocks via different pathways [7]. Based on our results, we may hypothesize that these clones may be highly responsive to proliferation stimuli (or may proliferate without stimuli). New pathways may be activated/inhibited by the co-expression of multiple CTAs, which may influence the effectiveness of salvage therapy, depending on the drug class (i.e. proteasome inhibitors). As the cumulative effects of the CTA cascade become clear in the future, the CTA expression profiles of patients at relapse may help to guide drug selection.
Overall, this small study has provided evidence that a coordinated expansion of stem cell clones progressively expressing more CTAs may occur during disease progression in MM (Fig. 4). We also demonstrated that a multiplexed CTA qRT-PCR assay combining PRAME/MAGEC1 and MAGEA3 or BAGE2 can be successfully used to monitor disease with greater sensitivity than is afforded by current biochemical methods. This method provides a clearer assessment of circulating disease based on progenitor cells as opposed to considering the PC component only. However, further studies are required to determine how this novel approach compares to current MRD methodologies for predicting relapse, confirm clonal evolution through sequential patient sample monitoring (flow cytometry), and ensure that the newer biological agents such as proteasome inhibitors do not delink CTA expression from malignant cells.
CTA biomarkers linked to advanced disease may allow for the monitoring of the clonal evolution of the disease (not possible with current MRD methods), which signals the development of more aggressive disease and may in future guide treatment choices as the effect of these proteins on cellular processes is established.


 XML Download
XML Download