

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
In several meta-analyses and cohort studies, fracture risk was shown to increase in patients with diabetes mellitus (DM) compared with controls without DM.12 Type 1 DM (T1DM) is associated with insulin deficiency and develops at a young age. Because insulin acts as an anabolic agent in bone, expected peak bone mass may not be achieved in patients with T1DM, resulting in low bone mineral density (BMD), which can increase the risk of fracture.3 Type 2 DM (T2DM) is associated with relative insulin deficiency and insulin resistance. Because patients with T2DM reportedly have higher BMD than controls without DM,1 increased fracture risk might be explained by decreased bone quality and increased risk of falls. Diabetic retinopathy, diabetic polyneuropathy,4 and hypoglycemic events5 are associated with increased risk of falls. Sarcopenia, which is low muscle mass and low muscle function, has been more frequently observed in patients with T2DM than in controls without DM.6 Sarcopenia is an important risk factor for falls7 and increases fracture risk.8
Bone formation and bone resorption markers are significantly decreased in patients with T1DM or T2DM compared with controls without DM.9 In a bone histologic study, bone formation was suppressed in patients with DM.10 In a Framingham high-resolution peripheral quantitative computed tomography study, higher cortical porosity and smaller cross-sectional area at the tibia were observed in patients with T2DM.11 Furthermore, the trabecular bone score in patients with T2DM was significantly lower than in subjects without DM.12 These low bone formation and bone remodeling rates, as well as abnormal bone microstructure in DM, are associated with bone quality deterioration, resulting in increased fracture risk. Advanced glycation end products (AGEs) are considered a mechanism of chronic diabetic complications and bone quality deterioration.13
AGEs are non-enzymatic glycation products of proteins, lipids, and nucleic acids with glucose through the Maillard reaction and the polyol pathway,14 and AGEs production increases in hyperglycemia and diabetes.15 AGEs accumulate in various tissues, such as bone, kidney, brain, and blood vessels. In patients with DM, accumulation of AGEs is associated with microvascular and macrovascular diabetic complications.16 The accumulation of AGEs in bone matrix, including abundant type 1 collagens, reduces the flexibility of collagen and bone strength.17 Although the effects of AGEs on bone cell dysfunction have been studied, evidence of their direct effects on bone cells is insufficient.
Therefore, in the present study, the direct effects of AGEs on differentiation and function of osteoblasts and osteoclasts were investigated using molecular analysis.
Go to : 
METHODS
Cell cultures and differentiation of osteoblasts and osteoclasts
MC3T3-E1 cells were not isolated osteoblasts but were used to evaluate the role of osteoblasts. The osteoblastic MC3T3-E1 cell line was obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). The MC3T3-E1 cells were cultured in an α-modified minimal essential medium (α-MEM; Welgene, Daegu, Seoul, Korea) supplemented with 10% fetal bovine serum (FBS; Gibco, Grand Island, NY, USA) and 1% penicillin/streptomycin (Gibco). The cells were maintained in osteogenic differentiation medium containing 10 mM β-glycerophosphate (β-GP; Sigma, St. Louis, MO, USA) and 50 μg/mL ascorbic acid (Sigma). Mouse bone marrow cells were obtained from femurs and tibias of four- to six-week-old male ICR mice (KOATECH, Pyeongtaek, Korea) and incubated in α-MEM complete media containing 10% FBS, 100 units/mL penicillin, and 100 μg/mL streptomycin on 100 mm culture dishes in the presence of 10 ng/mL macrophage colony-stimulating factor (M-CSF; PeproTech, Rocky Hill, NJ, USA) overnight. To obtain the bone marrow mononuclear (BMM) cells, which are cells of osteoclastic origin, bone marrow cells were cultured with 30 ng/mL M-CSF for two days without receptor activator of nuclear factor (NF)-κB ligand (RANKL; PeproTech). For osteoclast differentiation, bone marrow cells were cultured with 30 ng/mL M-CSF and RANKL 70 ng/mL for two days for polymerase chain reaction (PCR) and for three days for tartrate-resistant acid phosphatase (TRAP) staining.
AGEs treatment
Cultured cells were treated with 25 mM glucose and 25 mM mannitol was used as the osmotic pressure control. AGE-BSA was obtained from BioVision (Milpitas, CA, USA) and added to cultures simultaneously with cell differentiation factors, such as RANKL, ascorbic acid, or β-GP. AGEs were administrated at a concentration of 1, 10, or 100 ug/mL.
TRAP activity assay
To investigate osteoclast differentiation, cells were stained for TRAP activity. The cultured cells were washed with phosphate-buffered saline and fixed with citrate, acetone, and 3.7% formaldehyde for 1 minute. Leukocyte acid phosphatase kits (Sigma) were used according to the manufacturer′s instructions. Cells were washed with distilled water, and TRAP-positive multinucleated cells (MNCs) containing three or more nuclei and an actin ring were counted under a light microscope. A TRAP-positive MNC was considered a cell that could be an authentic osteoclast.
Alkaline phosphatase (ALP) and bone nodule formation assays
ALP and bone nodule formation assays were used to assess osteoblast differentiation. The early phase of osteoblast differentiation in MC3T3-E1 cells was assessed using ALP activity with ALP staining, and the late phase was assessed using bone nodule formation with Alizarin Red S staining. ALP activity was assayed by hydrolysis of p-nitrophenyl phosphate to p-nitrophenol using leukocyte ALP staining kits (86R-1 KT, Sigma). The measured absorbance was determined at 405 nm and compared with a p-nitrophenol standard titration curve. Enzyme activity was expressed as nM per dish and nM per microgram protein.
Matrix mineralization was assessed using Alizarin Red S staining in MC3T3-E1 cells. Cultures were fixed with 75% ethanol for 1 hour at 4°C, covered with Alizarin Red sodium monosulfonate (Sigma) for 30 minutes, and rinsed with water. Calcium deposits were destained with 10 mM sodium phosphate and 10% cetylpyridinium chloride buffer at pH 7.0, and absorbance was determined at 570 nm.
RNA extraction and gene expression analysis
Total RNA was isolated from cells using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). Cells were lysed directly in plate wells using 0.5 mL TRIzol per well. After chloroform extraction, total RNA was recovered from the aqueous phase and precipitated with equal volumes of isopropanol and diethylpyrocarbonate-treated water after a brief wash with 75% ethanol. Reverse transcription polymerase chain reaction (RT-PCR) was used to detect gene expression. RT-PCR was performed with 1 μg total RNA, oligodT (Bioneer, Daejeon, Korea), and reaction mixtures in AccuPower RT-PCR PreMix (Bioneer) at 42°C for 60 minutes. PCR primers were designed by Bioneer. Primer sequences and PCR conditions used in this study are listed in Table 1. The expression of osteoclast-specific genes such as RANK, TRAP, matrix metalloproteinases (MMP-9), cathepsin K, and nuclear factor of activated T-cells cytoplasmic 1 (NFATc1) was measured. Intercellular adhesion molecule 1 (ICAM-1) and lymphocyte function-associated antigen 1 (LFA-1)-α, β, which are necessary for osteoclast development, were measured. The expression of osteoblast-specific genes such as RUNX2, β-catenin, osterix, lysyl hydroxylase (LH), and lysyl oxidase (LOX) was measured.
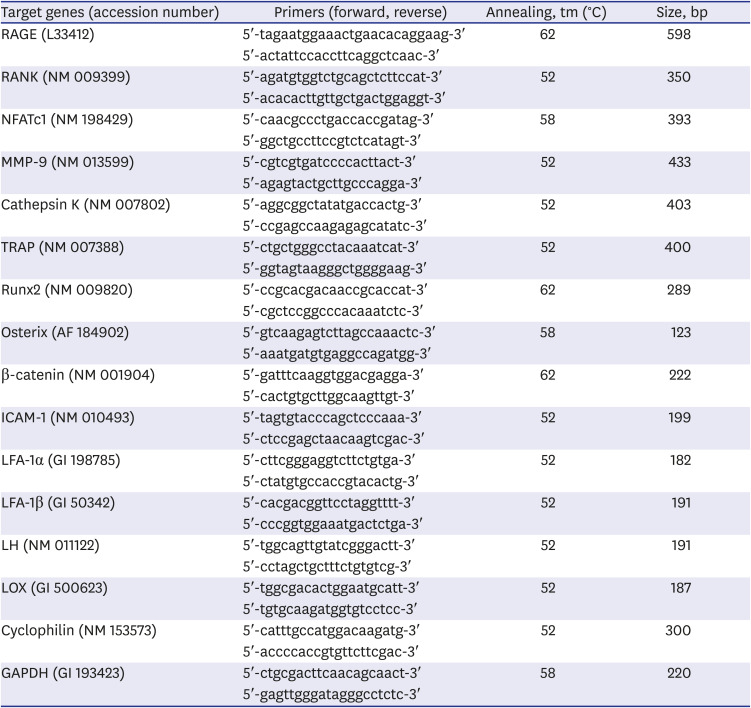
Table 1
Primer sequences and conditions for reverse transcription polymerase chain reaction
RAGE = receptor for advanced glycated end products, RANK = receptor activator of nuclear factor-κB, NFATc1 = nuclear factor of activated T-cells cytoplasmic 1, MMP-9 = matrix metalloproteinases-9, TRAP = tartrate-resistant acid phosphatase, ICAM-1 = intercellular adhesion molecule 1, LFA-1 = lymphocyte function-associated antigen 1, LH = lysyl hydroxylase, LOX = lysyl oxidase.
![]()
Western blot analysis
For isolation of total cell extracts, cells were lysed in RIPA lysis buffer (#9806, Cell Signaling Technology, Danvers, MA, USA) protein extract solution. Samples were centrifuged at 14,000 rpm × 10 minutes at 4°C. Protein concentration was determined using the BCA method (#23225, Thermo, Rockford, IL, USA). PVDF membranes (Millipore Co., Billerica, MA, USA) were incubated overnight at 4°C with primary antibodies at 1:1,000 in TBST. Antibodies against p-JNK, p-p38, p-ERK, p-AKT (Cell Signaling Technology), and β-actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA) were used for western blots.
Statistical analysis
Each experiment was performed in triplicate and results expressed as mean ± standard error of the mean. Statistical analysis was performed with IBM SPSS Statistics Software Version 18.0 (IBM, Armonk, New York, NY, USA). Analysis of variance was performed. Kolomogorov-Smirnov test was performed for the normality test. Significant differences (P < 0.05) between means were determined using Bonferroni test.
Ethics statement
All procedures used and the care of animals in this study were approved by the Animal Research Ethics Committee of Kyung Hee University Hospital at Gangdong (KHNMC AP 2016-004).
Go to :
RESULTS
Receptor for AGEs (RAGE) expression on cells
RAGE expression on MC3T3-E1, BMM cells, and marrow-derived MNCs was measured using RT-PCR. RAGE was expressed on all cells (Fig. 1).
 | Fig. 1RAGE expression in MC3T3-E1 cells, BMM cells, and marrow-derived MNCs using reverse transcription polymerase chain reaction. RAGE was expressed on all cells. The grouping of gels/blots cropped from different parts of the same gel.RAGE = receptor for advanced glycated end products, BMM = bone marrow mononuclear, MNC = multinucleated cell.
|
Inhibitory effects of AGEs on osteoclast and osteoblast differentiation
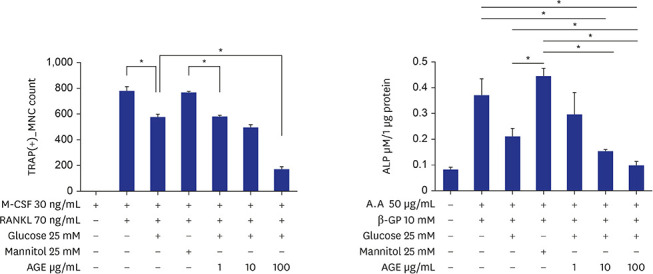
BMMs were incubated in 96-well plates (10,000 cells/well) for three days with 30 ng/mL M-CSF, 70 ng/mL RANKL, 25 mM glucose, 25 mM mannitol, and 1, 10, or 100 ug/mL of AGEs. Treatment with 25 mM glucose reduced the formation of TRAP-positive MNCs compared with the positive control. When 100 ug/mL AGEs were added under glucose treatment, formation of TRAP-positive MNCs further decreased significantly (Fig. 2A). To investigate ALP activity, MC3TC-E1 cells were incubated in 24-well plates (10,000 cells/well) for eight days with 50 ug/mL ascorbic acid, 10 mM β-GP, 25 mM glucose, 25 mM mannitol, and 1, 10, or 100 ug/mL of AGEs. Treatment with 25 mM glucose reduced ALP activity compared with positive control but without statistical significance. When 10 and 100 ug/mL AGEs were added under glucose treatment, ALP activity was further decreased significantly (Fig. 2B). To investigate bone nodule formation, MC3TC-E1 cells were incubated in 24-well plates (5,000 cells/well) for 14 days with 50 ug/mL ascorbic acid, 10 mM β-GP, 25 mM glucose, 25 mM mannitol, and 1, 10, or 100 ug/mL of AGEs. Treatment with 25 mM glucose and 10 or 100 ug/mL AGEs decreased bone nodule formation but without statistically significant difference (Fig. 2C).
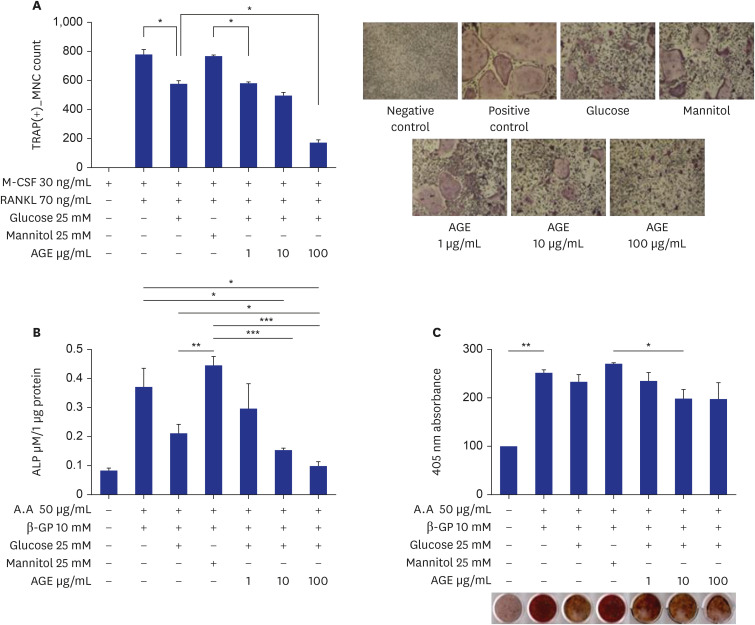 | Fig. 2Effects of AGEs on osteoclast and osteoblast differentiation. (A) TRAP-positive MNCs in RANKL-induced marrow macrophage cell cultures. Bone marrow mononuclear cells were incubated for three days with 30 ng/mL M-CSF, 70 ng/mL RANKL, 25 mM glucose, 25 mM mannitol, and 1, 10, or 100 ug/mL of AGEs (*P < 0.05). (B) ALP activities in MC3T3-E1 cell cultures. MC3TC-E1 cells were incubated for eight days with 50 ug/mL ascorbic acid, 10 mM β-GP, 25 mM glucose, 25 mM mannitol, and 1, 10, or 100 ug/mL of AGEs (*P < 0.05, **P < 0.01, ***P < 0.001). (C) Bone nodule formations in MC3T3-E1 cell cultures. MC3TC-E1 cells were incubated in 24-well plates (5,000 cells/well) for 14 days with 50 ug/mL ascorbic acid, 10 mM β-GP, 25 mM glucose, 25 mM mannitol, and 1, 10, or 100 ug/mL of AGEs (*P < 0.05, **P < 0.01).TRAP = tartrate-resistant acid phosphatase, MNC = multinucleated cell, M-CSF = macrophage colony-stimulating factor, RANKL = receptor activator of nuclear factor-κB ligand, AGE = advanced glycation end product, ALP = alkaline phosphatase.
|
Effects of AGEs on expression of osteoclast-specific genes and mitogen-activated protein kinase (MAPK) signaling pathways in RANKL-induced marrow cells
The effects of AGEs on the expression of several genes associated with osteoclast differentiation and the MAPK signaling pathways in response to RANKL were investigated. Expression of RANK, TRAP, MMP 9, cathepsin K, and NFATc1 decreased with increased AGEs (Fig. 3A). JNK and p38 as differentiation-related kinases and ERK and AKT as survival-related kinases in the MAPK pathway were measured in RANKL-induced marrow macrophages. JNK, p38, and AKT were suppressed after AGEs treatment (Fig. 3B); however, ERK expression was not different (Fig. 3B).
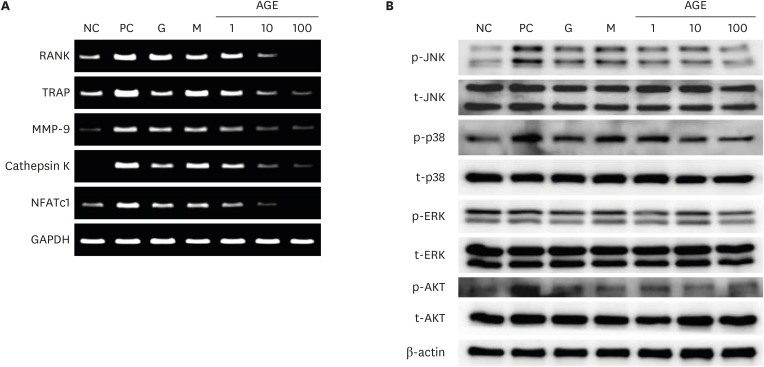 | Fig. 3Effects of AGEs on osteoclast-specific genes (A) and activation of the mitogen activated protein kinase signaling pathway (B) in RANKL-induced marrow macrophages. The osteoclast-specific genes such as RANK, TRAP, MMP-9, cathepsin K, and NFATc1 were measured by reverse transcription polymerase chain reaction. JNK, p38, ERK, AKT and β-actin were measured by western blots. The grouping of gels/blots cropped from different parts of the same gel.NC = negative control, PC = positive control, G = 25 mM glucose, M = 25 mM mannitol, AGE = advanced glycated end product, RANKL = receptor activator of nuclear factor-κB ligand, TRAP = tartrate-resistant acid phosphatase, MMP-9 = matrix metalloproteinases-9, NFATc1 = nuclear factor of activated T-cells cytoplasmic 1.
|
Effects of AGEs on ICAM-1 and LFA-1 expression
Expression of ICAM-1, LFA-1α, and LFA-1β were measured in RANKL-induced marrow macrophages. Expression of ICAM-1, LFA-1α, and LFA-1β decreased after AGEs treatment in a dose-dependent manner (Fig. 4).
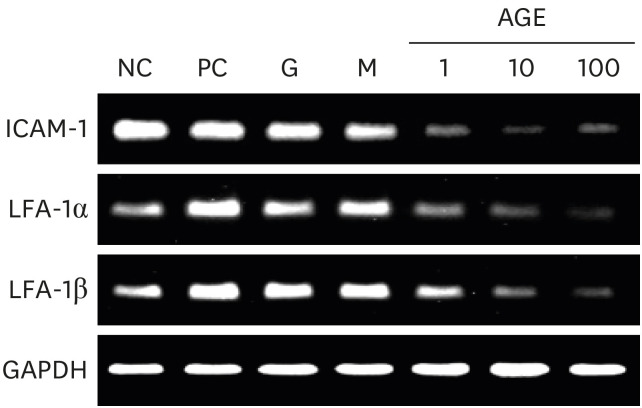 | Fig. 4Effects of AGEs on ICAM-1 and LFA-1 expression in marrow macrophages. Expressions of ICAM-1, LFA-1α, and LFA-1β genes were measured by reverse transcription polymerase chain reaction. The grouping of gels/blots cropped from different parts of the same gel.NC=negative control, PC=positive control, G=25mM glucose, M=25mM mannitol, AGE = advanced glycated end product, ICAM-1 = intercellular adhesion molecule 1, LFA-1 = lymphocyte function-associated antigen 1.
|
Effects of AGEs on expression of osteoblast-specific genes, LH, and LOX genes in MC3T3-E1 cells
AGE suppressed the expression of RUNX2, β-catenin, and osterix in a dose-dependent manner (Fig. 5A). AGE treatment decreased expression of LH and LOX genes (Fig. 5B).
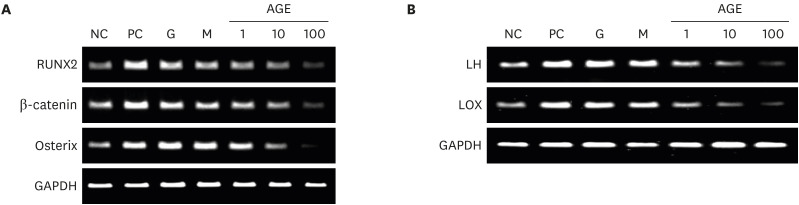 | Fig. 5Effects of AGEs on osteoblast-specific genes (A) and expression of LH and LOX genes (B) in MC3T3-E1 cells. The grouping of gels/blots cropped from different parts of the same gel. The osteoblast-specific genes, LH and LOX genes were measured by reverse transcription polymerase chain reaction.NC = negative control, PC = positive control, G = 25 mM glucose, M = 25 mM mannitol, AGE = advanced glycated end product, LH = lysyl hydroxylase, LOX = lysyl oxidase.
|
Go to :
DISCUSSION
The direct effects of AGEs on osteoblasts, osteoclast precursor cells, and osteoclasts were verified by confirming that all cells have receptors for AGEs. In the present study, AGEs suppressed differentiation and function of osteoblasts and osteoclasts in a dose-dependent manner. This finding indicates that longer duration of DM or more poorly controlled DM induces bone fragility. The ALP activity was observed higher in mannitol treatment than glucose treatment. It might have been seen prominently because MC3TC-E1 cell line used in this study had high differentiation activity through preparation with different medium composition.18
In the present study, the expression of NFATc1 and RANK was significantly decreased in high-glucose plus AGEs treatment compared with positive control, indicating that AGEs suppress osteoclast differentiation by inhibiting RANKL-RANK signaling via RANK expression reduction.
Reduced expression of JNK, p38, Akt, and NFATc1 indicates inhibition of three downstream pathways by RANKL/RANK signaling. The expression of TRAP, cathepsin K, and MMP9 was suppressed in this study. They are associated with the enzymes secreted from mature osteoclasts. Cathepsin K is a protease that catabolizes elastin, collagen, and gelatin. MMP9 is a type IV collagenase that degrades the extracellular matrix. This result indicates that AGEs inhibit not only differentiation but also the function of osteoclasts.
In this study, expression of ICAM-1 and LFA-1 in marrow macrophages was decreased after treatment with AGEs and consequently inhibited osteoclast formation. Cell-to-cell contact is necessary for osteoclast formation because osteoclasts form by cell fusion rather than endomitosis.19 ICAM-1 is expressed on osteoblasts, osteoclast precursors, and osteoclasts,20 and ICAM-1 expression can be regulated by intracellular signaling through NF-κB and JNK.21 ICAM-1 is a ligand for LFA-1,20 and ICAM-1 and LFA-1interactions are reportedly involved in cell-to-cell contact between osteoclast precursors and osteoblastic/stromal cells and between osteoclast precursors.21 The ICAM/LFA-1 interaction upregulates the expression of osteoclast-stimulating cytokines and stimulates RANK/RANKL signaling.22 Thus, AGEs inhibited osteoclast differentiation by inhibiting RANKL signaling and ICAM expression.
Expression of RUNX2, β-catenin, and osterix was decreased in MC3T3-E1 cells but essential for osteoblast formation. Decreased β-catenin expression also could change the osteoprotegerin (OPG)/RANKL ratio, but up-regulated RANKL expression would not mean enhanced bone resorption. Because RANK expression in osteoclast precursor cells was reduced after AGEs administration in this study, the change of OPG/RANKL ratio would not stimulate bone resorption. Because of other possible mechanisms related to AGEs, it should be considered as a whole. Osteoblasts synthesize type I collagen, which consists of 90% organic components of bone and determines bone strength. Enzyme-dependent collagen cross-linking depends on a combination of intracellular modifications of procollagen alpha chains by LH and on extracellular modifications by LOX.23 Decreased LH and LOX are associated with decreased bone quality.24 In the present study, AGEs induced bone fragility caused by low bone turnover and reduced bone quality.
In several previous studies, AGEs inhibited osteoblast differentiation and function. AGEs induced osteoblast apoptosis via TNF-α production, oxidative stress,25 endoplasmic reticulum stress,26 MAP kinase, and cytosolic apoptotic pathways.27 In addition, AGEs inhibited osteoblast differentiation by suppressing osterix expression.28 Conversely, results regarding the effects of AGEs on osteoclast differentiation are lacking. Tanaka et al.29 reported that AGEs inhibited human osteoclast differentiation through induction of IL-10 expression via NF-κB. In previous studies, AGEs suppressed osteoclast differentiation by increasing inflammatory cytokine levels. In contrast to other studies, AGEs inhibited osteoclast differentiation by reducing RANK, ICAM-1, and LFA-1 expression in this study. However, in other studies, AGEs enhanced osteoclastogenesis by RANKL mRNA upregulation and osteoclast-induced bone resorption.30 The results that AGEs suppressed LOX, causing structural abnormalities of bone and mineralization disorder, support our findings.31
The present study had several limitations. First, only a few genes associated with differentiation of osteoblasts and osteoclasts were analyzed, although several molecules are involved in multi-step differentiation. Second, differentiation of osteoclasts was measured, but activity was not directly measured. Third, AGEs could affect various organs and there are many confounding factors affecting bone. Therefore, results of this study on effects of AGEs on osteoblasts, osteoclasts, and type 1 collagen might be incomplete. How AGEs affect bone health should be investigated in future in vivo studies. However, this study is meaningful because the direct effects of AGE on osteoblasts and osteoclasts were analyzed. And this study is different from previous studies in that in analyzed the effect of AGE on not only osteoclast specific gene expression, but also the cell-to-cell contact, which is important for osteoclast differentiation, and analyzed bone quality related indicators such as LH and LOX.
In conclusion, AGEs inhibited differentiation and function of osteoblasts and osteoclasts, ICAM-1/LFA-1 interaction, and type 1 collagen cross-linking. AGE may induce bone fragility in patients with DM, through low bone turnover and decreased bone quality.
Go to :
 XML Download
XML Download