

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
The outbreak of respiratory coronavirus disease 2019 (COVID-19), caused by the severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2), is the most important public health challenge worldwide (1). SARS-CoV-2 is a positive-stranded, enveloped RNA virus, which can be zoonotically transmitted from animals to humans (2, 3). Bats comprise the most likely animal reservoir of SARS-CoV-2, but the presence of reservoirs in other animals has not been entirely excluded (4, 5, 6). The mortality rate of COVID-19 is approximately 3–4%, but the transmission rate of SARS-CoV-2 among humans is extremely high, presumably because of its air-droplet mode of transmission (7). COVID-19 has a wide range of symptoms, from mild-moderate to severe life-threatening infections; it may cause pneumonia, kidney failure, and death (7). Severe COVID-19 is associated with cytokine storm onset, which involves excessive and uncontrolled secretion of cytokines that may result in fatal sepsis and multi-organ failure (8, 9).
Cytokines and chemokines are protein messengers produced by immune cells, which have important roles in immunological and pathological responses during viral infections (10). Although coordinated cytokine induction is crucial for host immune response, dysregulated production is associated with immunopathology that can cause cytokine storm onset (11, 12, 13). Cytokines associated with COVID-19 include pro- and anti-inflammatory interleukins (ILs), chemokines, and interferons (10, 14). IL-17 is produced by T-helper 17 (Th17) cells; its signaling is associated with immune functions of barrier epithelial tissues and host defense against extracellular bacterial and fungal infections (15, 16, 17, 18). IL-17 includes unique structures composed of IL-17A and IL-17F (17); it is an inducer of antimicrobial proteins, various chemokines, acute-phase response mediators, and inflammatory functions (16, 19). Recent studies have found increased inflammatory responses in COVID-19 patients because of IL-17 overproduction (20, 21). In this review, we discuss the characteristics of IL-17A, its relevance to COVID-19, and the potential for IL-17A-based therapeutics.
Go to : 
OVERVIEW OF IL-17 AND ITS SIGNALING NETWORK
Cytokines, combined with specific receptors, have pleiotropic roles in innate and adaptive immune responses that involve complicated intracellular signal pathways (22). Cytokine dysregulation can cause pathological effects in the host immune system and tissues (23). This section provides a brief overview of IL-17 in terms of biological function and signaling pathways, including JAK and STAT3.
Biological functions of IL-17
IL-17, produced mainly by Th17 cells (24), has crucial roles in adaptive immunity and inflammatory responses during infection (24). IL-17 is a member of the inflammatory cytokine family that can exert both host-protective and pathological effects (21). The IL-17 family consists of six structurally related members: IL-17A, IL-17B, IL-17C, IL-17D, IL-17E (IL-25), and IL-17F (18). IL-17A is the prototype of the IL-17 cytokine family; it is co-expressed along with IL-17F by Th17 cells (25). IL-17A is also produced by various hematopoietic cells, including CD8+ T cells, invariant natural killer T cells, gamma delta T cells, neutrophils, and type 3 innate lymphoid cells (26, 27). IL-17A is an essential cytokine involved in host defense against extracellular bacterial and fungal infections (15, 17, 18). However, it also contributes to pathologically excessive pro- inflammatory responses related to the development of autoimmunity, tumors, and other inflammatory diseases (15, 28) (Fig. 1).
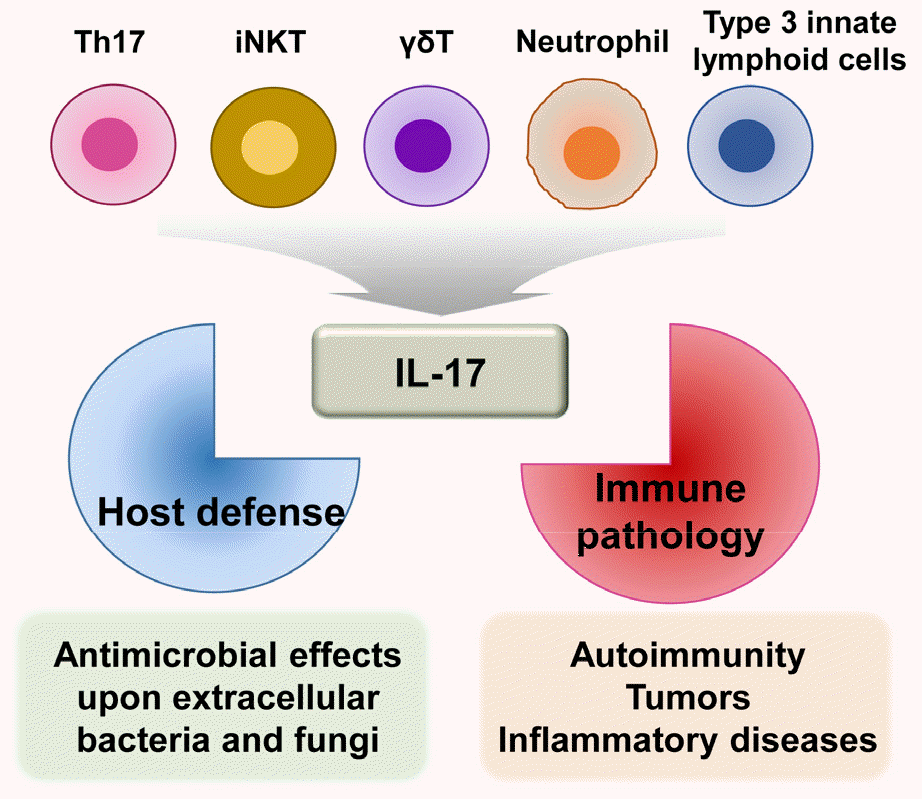 | Fig. 1IL-17 production and function. IL-17 is mainly produced by Th17 cells and other cell types, including CD8+ T cells, invariant natural killer T cells, gamma delta T cells, neutrophils, and type 3 innate lymphoid cells. IL-17 has a dual function: host defense against extracellular bacterial and fungal infections, as well as pathologic inflammation involved in various diseases (e.g., autoimmune disorders, tumors, and inflammatory diseases).
|
IL-17 binds to receptors to induce an immune response specific to the target cells (15). The IL-17R family consists of five members (IL-17RA, IL-17RB, IL-17RC, IL-17RD, and IL-17RE); IL-17RA is the principal member (25, 27). The IL-17/IL-17R interaction is required for inflammatory responses through various signaling molecules and transcriptional factors, including nuclear factor-κB, Janus kinase (JAK)–STAT, and tumor necrosis factor receptor-associated factor-6 (15, 29). Intracellular signaling pathways involving the JAK–STAT3 interaction are discussed in detail in the following section.
JAK–STAT signaling pathway and STAT3
The JAK–STAT pathway is a representative cytokine signaling pathway that mediates messages from membrane receptors to the nucleus (30, 31). The JAK–STAT signaling pathway is composed of three classes of proteins: tyrosine kinase-related receptors, JAK enzymes, and signal transducer and activator of transcription (STAT) transcription factors (32). JAKs are phosphorylated in the presence of cytokine ligands, thus creating STAT docking sites. STAT molecules are phosphorylated and dimerized by JAK, then transported to the nucleus to regulate expression of cytokine-responsive genes in the nuclear membrane (33).
The JAK–STAT signaling pathway is critical for transducing signals of various cytokines and growth factors (e.g., IL-6, granulocyte-macrophage colony-stimulating factor, growth hormone, epidermal growth factor, platelet-derived growth factor, and interferons) in various cell types (30). IL-6, IL-1, and IL-23 activate STAT3, a transcriptional factor required for Th17-cell differentiation (34). Retinoic acid-related orphan receptor (ROR)γt is another transcription factor crucial in Th17-cell development. Th17-cell differentiation is activated by IL-1β, IL-6, and transforming growth factor-β; it is maintained by IL-23 (20, 34). Thus, multiple cytokines and transcriptional factors are involved in IL-17 production by Th17 cells.
STAT3 activates RORγt transcription, which regulates Th17-cell differentiation (20). The STAT3–RORγt interaction is important for Th17-cell differentiation (24). Th17-cell survival and differentiation require IL-23 and IL-21, which are produced by dendritic and T cells, respectively (24). IL-6 regulates Th17 cells through a positive feedback loop that amplifies the expression and activation of IL-17 and STAT3 (35). Th17-cell differentiation is promoted by the binding of IL-6 and transforming growth factor-β to naive CD4+ T cells (36). However, Th17-cell differentiation does not require STAT4 or STAT6, which are transcription factors that regulate Th1 and Th2 reactions, respectively (37). Indeed, STAT3 is essential for Th17-cell differentiation (38). Because STAT3C is an active STAT3 variant, its overexpression increases the number of IL-17-producing lymphocytes. This results in increased RORγt expression in CD4+ T lymphocytes (39). In contrast, STAT3 deficiency in hematopoietic stem cells, especially CD4+ lymphocytes, results in a sharp decrease in the number of Th17 lymphocytes (39, 40). Together, STAT3 and RORγt are crucial for the activation of IL-17-producing Th17 cells. Fig. 2 summarizes the differentiation of Th17 cells and the related cytokine network. Details of IL-17/IL-17R signaling pathways are beyond the scope of this review; they have been discussed in several recent reviews (15, 27, 41). The next section describes the roles of IL-17 and Th17-related risk factors in the pathophysiology of COVID-19.
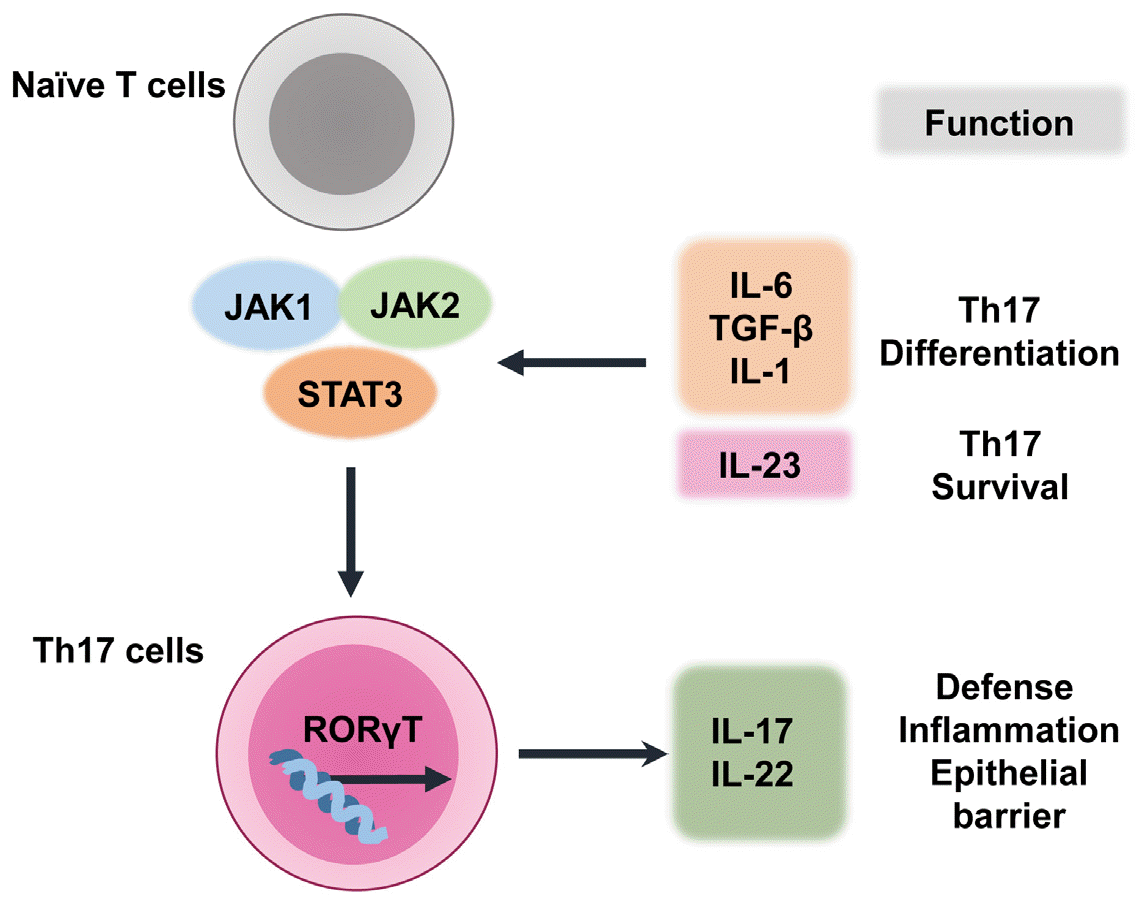 | Fig. 2Th17-cell differentiation and related cytokine network. In naive T cells that are committed to differentiate into Th17 cells, multiple cytokines (e.g., IL-6, IL-1, transforming growth factor-β, and IL-23) activate STAT3, which has a critical role in Th17-cell differentiation. RORγt is a crucial transcription factor in Th17-cell development, and STAT3 is a key factor in the transcriptional activation of RORγt. Finally, Th17-cell survival and differentiation are assisted by IL-23.
|
Go to :
ROLE OF IL-17 IN COVID-19
IL-17 and pathological responses in COVID-19 patients
Previous studies have reported upregulation of Th17 cells in COVID-19 patients (42, 43). IL-17 is involved in the pathogenesis of acute respiratory distress syndrome through the enhancement of neutrophil infiltration into the lungs (44, 45, 46). Increased IL-17A signaling and upregulation of Th17 or regulatory T cells (Treg) are positively correlated with acute respiratory distress syndrome severity in Middle East respiratory syndrome-related coronavirus and respiratory syncytial virus infections (47, 48). Similarly, Th17-cell overactivation and exaggerated cytotoxic effects of CD8+ T cells are at least partly responsible for severe immune injury in COVID-19 patients (43). A recent study showed that COVID-19 patients with pneumonia exhibited exhausted T-cell profiles with upregulated Th17 responses (49). IL-17A is associated with lung inflammation and poor clinical outcomes in viral acute respiratory distress syndrome (45). IL-17A signaling amplifies pathological inflammation through the induction of pro-inflammatory cytokines, such as IL-6 and IL-1; thus, it is considered a promising target for adjunctive acute respiratory distress syndrome treatment in COVID-19 patients (44). Despite a known association between IL-17 and acute pulmonary injury in COVID-19 patients, the underlying functional mechanisms are poorly understood.
There is substantial evidence to support an interaction between IL-17 and angiotensin-converting enzyme 2 (ACE2), an entry receptor for SARS-CoV-2 (50, 51, 52, 53). Murine models have demonstrated a relationship between pulmonary human ACE2 expression and COVID-19 development, along with a pro-inflammatory immune response (54). In a murine model of severe bacterial pneumonia, recombinant ACE2 has been shown to inhibit IL-17A-mediated STAT3 activation and decrease pulmonary neutrophil infiltration. This suggests a protective role for ACE2 in bacterial lung infections (55). Although the relationship between ACE2 expression and COVID-19 severity is complex, ACE2 is an important regulator of IL-17A production (53, 56). IL-17A-mediated pulmonary inflammation in COVID-19 patients may lead to an increased number of alveolar neutrophils and organ dysfunction (20); therefore, future research should address pathogenic IL-17A responses and factors related to IL-17A signaling, which can affect the clinical severity of COVID-19. Understanding the mechanism of increased IL-17A production in various tissues during SARS-CoV-2 infection may provide novel strategies for treatment using IL-17-based drugs.
Risk factors and Th17 responses in COVID-19
Risk factors for COVID-19 include age, obesity, sex, cardiovascular diseases, chronic kidney disease, hypertension, and diabetes mellitus (57, 58, 59, 60, 61). These risk factors are known to upregulate Th17 responses and IL-17A signaling (20). It has been suggested that pro-inflammatory responses (e.g., increased IL-17A levels) contribute to lung damage and hypercoagulability in patients with severe COVID-19 (62). Although the effects of these risk factors on IL-17A production and Th17 responses are poorly understood, their relationship supports the possibility for use of IL-17A-targeting modalities in the management of severe COVID-19 patients with high risk of mortality. In this section, we have explored the effects of some risk factors on IL-17 levels and pathological responses in COVID-19.
Age
COVID-19 patients aged ≥ 80 years have a 20-fold greater risk of mortality, compared with such patients aged 50–59 years (63). Although the age-related alterations in Th17 responses are not well understood, there is evidence for upregulated differentiation of IL-17-producing effector cells from naive CD4+ T cells in healthy volunteers aged > 65 years (64). A recent study showed that elderly individuals had increased Th17 responses (including upregulated RORγt expression and IL-17 levels in peripheral blood), compared with middle-aged and young individuals (65). IL-17 treatment of human umbilical vein endothelial cells has been shown to cause elevated endothelin-1 and vascular cell adhesion protein-1 levels. This suggests a relationship between Th17 responses and endothelial cell senescence (65). Another study reported that transcriptional activation of IL-17-, IL-17F-, IL-22-, and IL-17-producing CD4+ T cells significantly increased with age in humans and mice (66). Increased IL-17 and Th17 responses were also reported in aged mice due to increased mRNA expression of human-homologous RORγt (66). Adoptive transfer of naive T cells from aged mice leads to the development of colitis in RAG−/− mice, suggesting a role for Th17 in the pathogenesis of inflammatory disorders (66). Moreover, the balance between Th17 (CD4+ IL23 receptor(R)+) and Treg (CD4+ Foxp3+) cells is dysregulated in old age, resulting in Th17-cell predominance (67). Although these studies suggest that immune senescence is closely related to increased Th17 responses and IL-17 production, the COVID-19-specific roles of age-related alterations in Th17–Treg balance remain poorly understood.
Cardiovascular diseases and hypertension
SARS-CoV-2 spike protein binds to ACE2, an important component of renin–angiotensin and cardiovascular systems, to facilitate viral invasion into host cells (68). Numerous studies have demonstrated a link between COVID-19 and cardiovascular diseases (69, 70, 71, 72). SARS-CoV-2 infection triggers endothelial injury and dysfunction, leading to increased mortality in COVID-19 patients (56, 68, 73). Although the mechanisms underlying myocardial injury are largely unknown, Th17 dysregulation may contribute to COVID-19 outcomes in patients with comorbid cardiovascular diseases and hypertension.
IL-17 is involved in angiotensin II-mediated hypertension and cardiovascular dysfunction (74); it comprises a therapeutic target for cardiovascular diseases. Th17-cell differentiation causes organ damage in hypertensive mice (75), whereas genetically IL-17A-deficient mice have low blood pressures because of Th17–Treg imbalance (74). Patients with hypertension have increased numbers of Th1 and Th17 cells, as well as increased levels of related cytokines (76). Based on these reports, hypertension is presumed to be closely associated with increased Th17 response and Th17–Treg imbalance, thereby contributing to COVID-19 severity.
Obesity
Obesity has been suggested to cause chronic inflammation, thereby leading to Th17-related metabolic and autoimmune diseases (77, 78). An experimental study found greater numbers of IL-17-secreting CD4+ T cells in obese mice than in lean mice (78). The increased Th17 responses associated with obesity are dependent on IL-6 production; they are linked to autoimmune disease susceptibility (78). Altered regulation of intracellular metabolism, including fatty-acid metabolism, might result in obesity-related Th17 responses (79). Studies have shown higher IL-17 and IL-23 levels in obese women than in lean women (80). Based on these findings, IL-17 pathway activation might be amplified in obesity, potentially leading to adverse clinical outcomes in patients with COVID-19 (81). The mechanisms by which obesity induces Th17 inflammatory responses are unclear. Th17–Treg imbalance may cause dysregulated immune homeostasis because lean adipose tissues are rich in Tregs, which control tissue inflammation (82). Notably, acetyl-CoA carboxylase 1 (ACC1) was found as a key regulator of Th17 cell differentiation through regulation of RORγt binding to the Il17a gene (83). Further studies are needed to clarify the molecular mechanisms underlying Th17-cell increase and Treg-cell decrease, as well as the roles of obesity-related metabolic changes.
Diabetes mellitus
Diabetes mellitus and hyperglycemia are considered predictors of poor prognosis in COVID-19 (84, 85). Hyperglycemia is linked to dysregulated innate immunity involving upregulated expression of Toll-like receptors and defective neutrophil function (86). In addition, hyperglycemia leads to protein glycosylation, altered complement function, and phagocytosis inhibition (86). Therapies for COVID-19 may lead to poor glycemic control because of virus-triggered β-cell damage and increased insulin resistance through exaggerated inflammatory cytokine production (87). Thus, a vicious cycle might arise involving diabetes mellitus and COVID-19 (87).
Th17 dysregulation is likely to contribute to complex interactions between COVID-19 and diabetes mellitus. Patients with types 1 and 2 diabetes have increased Th17–Treg imbalances and Th17-cell activation, compared with controls (20, 88, 89, 90). Diabetic patients have lower levels of miR-146a, which regulates Th17-cell differentiation by inhibiting the expression of cytokines such as IL-6 and IL-21 (91). In addition, increased pro-inflammatory cytokine signaling and poor glycemic control in diabetic patients are associated with Th17-response dysregulation (91, 92). These studies suggest that IL-17-targeting therapeutics may be useful in the treatment of diabetic COVID-19 patients.
Sex
Several studies have suggested sex-differences in the clinical outcomes of COVID-19. A recent meta-analysis showed a higher prevalence of COVID-19 in men (93). Smoking and alcohol consumption were also more prevalent in men, and they have been linked to increased Th17-cell frequencies (94, 95). Furthermore, severe COVID-19 was more common in men than in women, although robust analyses are needed to further evaluate these sex differences (96, 97, 98). Male COVID-19 patients had higher plasma inflammatory cytokine levels, such as IL-8 and IL-18, whereas female patients had upregulated T-cell activation. Male COVID-19 patients with severe disease also exhibited a negative correlation between a poor T-cell response and age (97). However, the molecular mechanisms by which sex is associated with COVID-19 severity are unknown.
There is evidence to suggest that estrogen and progesterone have roles in the regulation of Th17 responses (99). Estrogen deficiency increases Th17-cell differentiation and lowers the number of Treg cells. This is partly because of the inhibitory effects of estrogen on RORγt activation, which interfere with Th17-cell differentiation (100, 101). In addition, estradiol treatment impairs antigen presentation and IL-23 production by dendritic cells, thereby inhibiting Th17 immune responses (102). In an experimental viral myocarditis model, Th17-cell responses were significantly greater in male mice than in female mice, indicating sex differences in terms of susceptibility to Coxsackievirus B3 infection (103). Progesterone and estrogen inhibited the production of Th1/Th17 inflammatory cytokines, although they increased the production of anti-inflammatory cytokines in human peripheral blood mononuclear cells from women with recurrent spontaneous miscarriages (104). Further studies are needed to clarify whether men have increased Th17 responses, which may increase COVID-19 susceptibility and severity. Additionally, estradiol and progesterone levels may influence COVID-19-related cytokine storm outcomes via enhanced immune tolerance (105). A greater understanding of the role of sex in COVID-19 may contribute to the development of potential sex-based therapeutic approaches.
Acute and chronic kidney diseases
Several studies have suggested that COVID-19 risks and outcomes may be related to chronic kidney disease and kidney transplantation (106, 107, 108). In addition, COVID-19 may be complicated by acute kidney injury, leading to a greater mortality risk (109, 110). Although the underlying mechanisms are largely unknown, Th17 responses might contribute to COVID-19 outcomes in patients with comorbid chronic kidney diseases. An association between common single nucleotide polymorphisms in IL17RA and a risk of renal disease has been demonstrated (111). The homozygous single nucleotide polymorphism rs4819554 AA in IL17RA increases IL17RA protein expression and Th17 responses, which might contribute to renal damage and impaired kidney function (111). IL-17A is involved in numerous pathological conditions related to the development of acute and chronic renal diseases (112, 113, 114). Chronic kidney disease patients often have Th17–Treg imbalances and increased IL-17A responses (112). Table 1 summarizes potential mechanisms by which several risk factors may alter Th17 responses in COVID-19. Further studies are needed to clarify the implications of renal disease in COVID-19 via altered Th17 responses involving IL-17A hyperactivation.
Table 1.
Potential mechanisms by which the risk factors influence altered Th17 responses in COVID-19
| Risk factors of COVID-19 | Study model | Pathologic outcomes in various disease conditions | Reference |
|---|---|---|---|
| Aging | Human PBMCs | The increased Th17 differentiation from naïve CD4+ T cells in healthy eldery group | (64) |
| Human endothelial cells | IL-17 leads to the elevation of ET-1 and VCAM-1 levels | (65) | |
| C57BL/6 mice | Increased Th17 cytokines and frequncies in aged mice | (66) | |
| Cardiovascular disease | IL17-/- mice | ACE2 enhances aortic T cell infiltration with increased IL-17 and other inflammatory cytokines | (74) |
| C57BL/6 mice | ACE2 increases Th17 cells through upregulation of SGK1- FoxO1 pathway | (75) | |
| Human PBMCs | Increased Th17 cells and IL-17 levels in hypertensive patients | (76) | |
| Obesity | ACC1-/- mice Human PBMCs | ACC1 increases Th17 diffrentiation through RORγt activation | (83) |
| Type 1 diabetes | Human PBMCs | Elevated Th17 responses positively correlate with CRP | (89) |
| Type 2 diabetes | Human PBMCs | Increased Th17 responses positively correlate with nephropathy | (90) |
| Alcohol | RORγt-/- mice | Th17 drives alcohol-associated gut inflammation and the development of ALD | (94) |
| Smoking | Human PBMCs | Increased CCR6+ Th17 cells and VEGFα secretion | (95) |
| Progesterone deficiency | Human PBMCs | Progesterone decreases STAT3-IL-6 signaling | (99) |
| Estrogen deficiency | BALB/c mice | Ovariectomized mice increases RORγt, STAT3, and IL-17 | (101) |
| Male | BALB/c mice | Upregulated Th17 responses in Coxsackievirus B3-infected male mice increases susceptibility of myocarditis | (103) |
| Kidney disease | Human PBMCs | Increased Th17 frequencies and IL-17 production from effector memory T cells in end-stage renal disease patients | (113) |
| Human PBMCs | Enhanced Th17 diffrentiation and IL-17 levels positively correlate with phosphate levels in chronic hemodialysis patients | (114) |
ACC1, acetyl-CoA carboxylase 1; ACE2. angiotensin-converting enzyme 2; ALD, alcohol-associated liver disease; COVID-19, coronavirus disease 2019; CRP, C-reactive protein; ET-1, endothelin-1; FoxO1, forkhead box O1; IL, interleukin; PBMC, peripheral blood mononuclear cell; RORγt, retineic-acid-receptor-related orphan receptor gamma T; SGK1, serum/glucocorticoid regulated kinase 1; Th17, T helper 17; VCAM-1, Vascular cell adhesion molecule-1. VEGFα, vascular endothelial growth factor α
![]()
IL-17-targeting therapeutics in COVID-19
Despite some side effects, monoclonal antibodies that interact with the IL-17 signaling pathway (e.g., secukinumab, ixekizumab, and brodalumab) have been used in therapeutic trials for several IL-17-related autoimmune diseases, including psoriatic diseases and rheumatic diseases (52, 115, 116). The success of IL-17 antagonists in the treatment of autoimmune diseases has encouraged their use in the treatment of COVID-19. In particular, JAK inhibitors, including baricitinib, ruxolitinib, and fedratinib, showed effective anti-inflammatory responses in COVID-19 patients through suppression of the JAK–STAT pathway (117). A recent experimental study showed that ex vivo treatment of whole-blood samples from COVID-19 patients with baricitinib led to a significant decrease in viral spike-specific cytokine release (118). Another study showed that fedratinib, a Food and Drug Administration-approved Janus kinase 2 (JAK2) inhibitor, suppressed the Th17 pathway; this reduced mortality and ameliorated the cytokine storm that occurs in COVID‐19 patients (42). In addition, ruxolitinib, a JAK1 and JAK2 inhibitor, is currently under investigation (ChiCTR2000029580) for potential therapeutic application involving interference with the inflammatory response in COVID-19 (119). Furthermore, considering the close relationship between IL-17 and IL-6, a combined approach to block these cytokines has been proposed as a potential treatment for COVID-19 (26). However, caution is needed because IL-17 deficiency can contribute to small bowel pathology and cytokine release syndrome through an interferon-γ–STAT1-mediated inflammatory pathway (120). Further studies are necessary to clarify whether IL-17-targeting therapeutics can reduce the severity of COVID-19.
Go to :
CONCLUSION
IL-17A is a key inflammatory cytokine, mainly produced by Th17 cells, which leads to the activation of various pro- inflammatory cytokines and chemokines. This causes lung injury in COVID-19; it also contributes to other inflammatory diseases. Emerging data suggest a pivotal role for IL-17 in pathological inflammatory responses, including severe COVID-19. In addition, IL-17A has been linked to risk factors for poor COVID-19 outcomes (e.g., immune senescence, cardiovascular diseases, obesity, diabetes mellitus, sex, and kidney injury). However, the underlying molecular mechanisms are largely unknown. Although several studies have indicated that excessive IL-17 production is related to the development of acute respiratory distress syndrome in patients with COVID-19, further studies in larger populations are needed to determine the IL-17 levels in sera, plasma, and bronchoalveolar lavage fluids from patients with different clinical phenotypes. This would clarify the usefulness of IL-17-targeting therapeutics in subgroups of patients with IL-17 hyperactivation. The success of IL-17-targeting therapeutics (monoclonal antibodies and JAK–STAT signaling inhibitors) in several autoimmune diseases encourages their potential use in the treatment of COVID-19. Preclinical and clinical data must be accumulated to identify the factors that determine whether IL-17-targeting therapeutics should be used alone or in combination with other drugs or monoclonal antibodies. These translational studies based on IL-17 and Th17 functions suggest the potential for development of improved COVID-19 treatment strategies.
Go to :
 XML Download
XML Download