

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Pancreatic cancer is the second most prevalent digestive system cancer after colorectal cancer, with an estimated 56,000 cases and 45,000 deaths in the United States in 2019 [1]. Worldwide, there were more than 450,000 cases of pancreatic cancer and more than 430,000 deaths in 2018 [2].
Adenocarcinoma of the pancreas is characterized by a low tumor mutation burden molecularly, with most cases in the Cancer Genome Atlas (TCGA) pancreatic cancer study having less than 50 mutations, a minority having 50 to 80 mutations, and only a few cases having more than 80 mutations [3]. However, a handful of cancer-associated genes display recurrent mutations that can be used to characterize the pathogenesis of the disease. These include mutations in the KRAS oncogene observed in the preponderance of cases ranging from 65% to 90% in different series and mutations of the TP53 gene encoding for p53 protein, a major tumor suppressor. In TCGA, about two thirds (65.4%) of cases bear mutations in KRAS, which is the most frequently mutated gene in pancreatic cancers [3]. However, in the more extensive published series of the MSK-IMPACT study a pancreatic adenocarcinoma subset with 384 patients and a pancreatic adenocarcinoma cohort of the GENIE project with 3,004 patients, the frequency of KRAS mutations approached 90% [4,5]. TP53 is the most frequently mutated tumor suppressor. Mutations of TP53 have been found in about 60% of cases in TCGA and in about 70% of pancreatic adenocarcinoma subsets of the MSK-IMPACT study and the GENIE project. A cell cycle inhibitor p16 encoded by the CDKN2A gene and a transforming growth factor beta pathway signal transducer SMAD4 are two other commonly mutated tumor suppressors in pancreatic cancer. Their mutations have been found in about 20% of cases [3,5]. In addition, the p16 locus at chromosome 9p, which also encodes for the p53 positive regulator p14ARF, is deleted in 10% to 25% of pancreatic cancer cases. Beyond these molecular abnormalities, no other oncogenes or tumor suppressors are recurrently altered in pancreatic cancers in more than 6% of cases. This fact is also mirrored by a low total mutation burden and a low score for aneuploidy that characterize this disease.
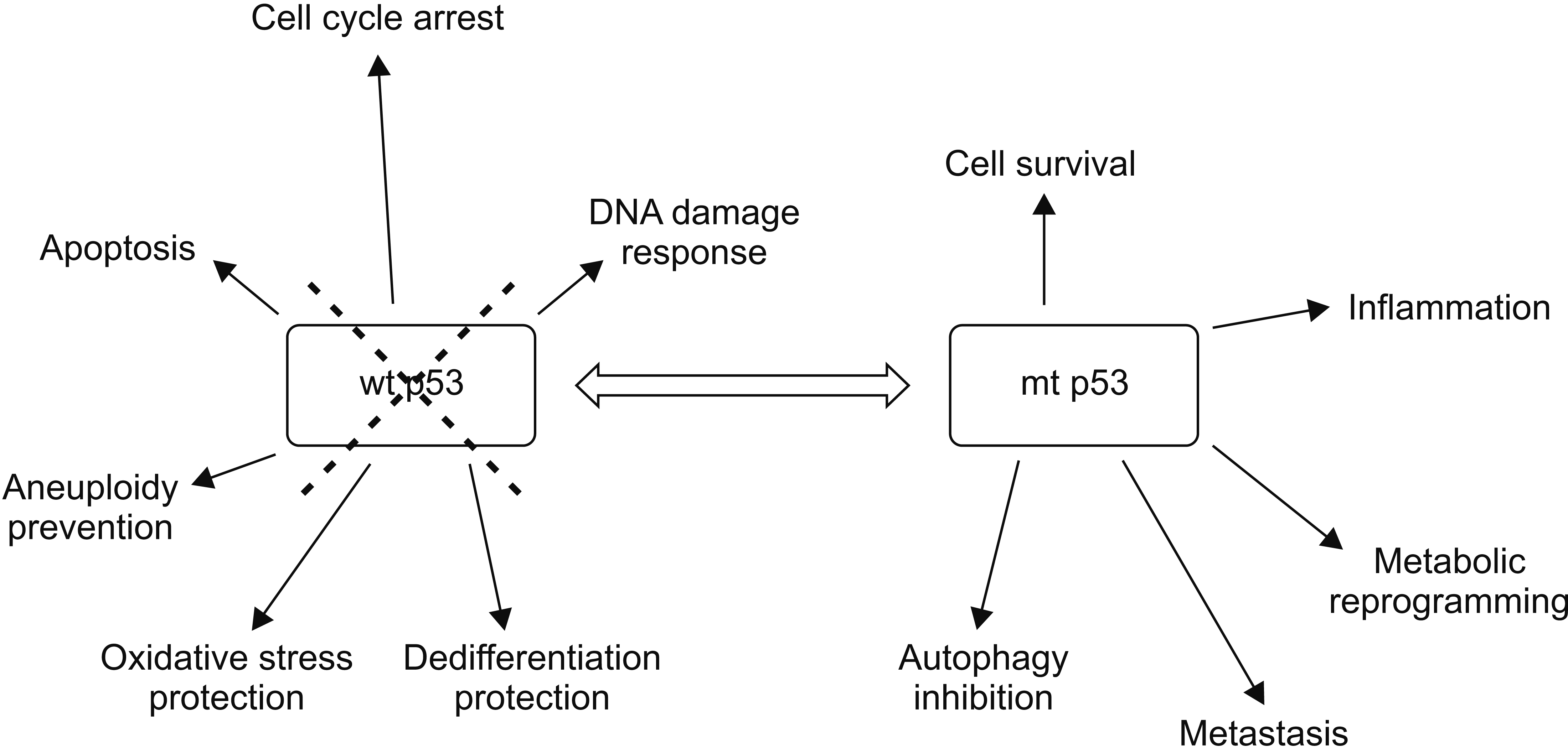
About two-thirds of p53 mutations in pancreatic cancer are missense mutations, while the remaining third are truncating mutations [4]. The specific type of mutation may be of significance as point mutations can produce mRNAs that are translated to proteins that can interfere with the function of their wild-type counterparts. By contrast, truncating mutations produce mRNAs that are degraded due to haplo-insufficiency of the wild-type allele [6]. Implications of the presence of mutant p53 extend beyond interference with the normal function of its wild-type protein given that the mutant protein acquires gain of functions that are tumor-promoting (Fig. 1) [7]. However, the most well-agreed upon effects of TP53 mutations are derived from the absence or decrease of physiologic functions of its normal protein [7]. These functions include cell cycle arrest, apoptosis promotion, DNA damage response triggering, maintenance of cell polarity, and maintenance of genomic stability [8,9].
The current paper will discuss the landscape of mutations of the gene encoding for p53 in pancreatic adenocarcinomas and juxtapose these cancers having p53 mutations with pancreatic cancers without such mutations.
MUTATIONS OF p53 IN PANCREATIC CARCINOGENESIS: FROM PRECURSOR LESIONS TO INVASIVE CANCER
Precursor lesions and role of risk factors
KRAS is a main oncogene that drives pancreatic cancer initiation. It is present in precursor lesions of the disease, including intraductal papillary mucinous neoplasms (IPMN) and pancreatic intraepithelial neoplasia (PanIN) [10]. p53 mutations can develop later. They are observed in some PanIN lesions, but not in IPMN. KRAS mutations are only weakly or not at all associated with pancreatic cancer cases in smokers [11,12]. However, this relationship has been difficult to establish or refute because the prevalence of KRAS mutations in pancreatic cancer is almost universal. In addition, the number of cases is small in most studies examining subjects with pancreatic cancer. Likewise, TP53 mutations that are prevalent in subsequent steps of pancreatic carcinogenesis do not seem to correlate with the smoking history of patients [11,13]. This contrasts with the total mutation burden of pancreatic cancers that appears to be higher in smokers [13]. It also contrasts with studies on non-small cell lung cancer (NSCLC), a prototypical smoking-associated cancer where an increased prevalence of KRAS and TP53 mutations is observed in smokers. The total mutation burden in pancreatic cancer is also generally much lower than that in NSCLC, a fact that correlates with low responses of pancreatic adenocarcinoma to immunotherapies. However, immunotherapies with checkpoint inhibitors have become part of the standard treatment for NSCLC, especially for cancers with different levels of positive expression for the PD-L1 receptor [14].
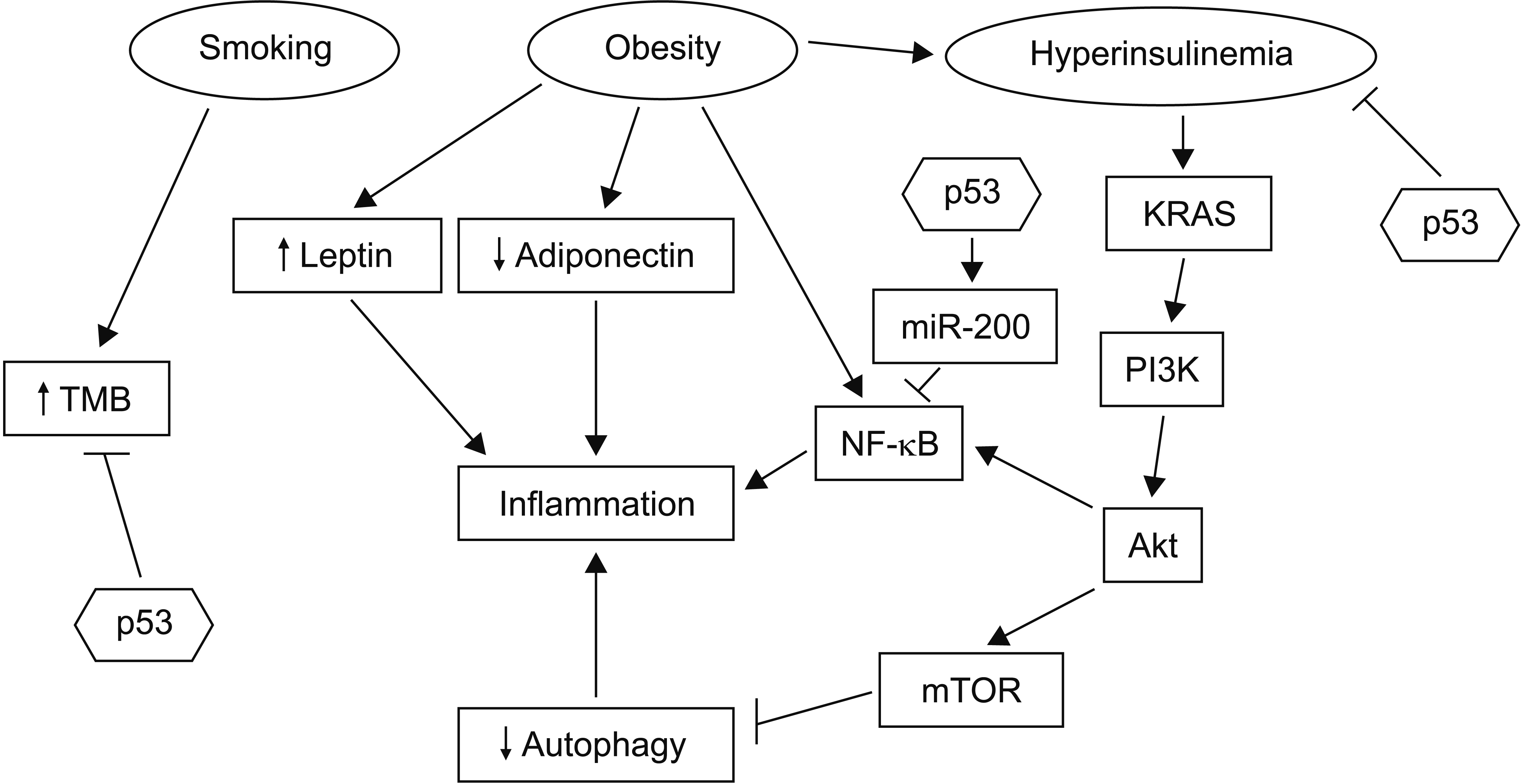
The association of pancreatic cancer with obesity, another risk factor, is pathophysiologically linked to inflammation, chronic pancreatitis, and dysfunction of autophagy [15]. Obesity can cause upregulation of leptin, a proinflammatory protein, and downregulation of anti-inflammatory adiponectin (Fig. 2). In addition, proinflammatory cytokines can be induced by the activation of transcription factor NF-κB. Obesity can inhibit autophagy by activating signals of sufficient nutrient availability through the PI3K/Akt/mTOR pathway [15]. Inhibition of autophagy can further promote inflammation through the activation of alternative clearance pathways of defective organelles that are inflammasome dependent [16]. Moreover, although autophagy may promote cell survival in established cancers, the inflammation-promoting effect of autophagy dysfunction is important for the maintenance of early pancreatic neoplastic lesions [17,18]. p53 can guard against this vicious cycle through several mechanisms, including inhibition of NF-κB signaling by inducing miR-200 family micro-RNAs and inhibiting the JAK-STAT3 pathway [19]. Active STAT3 signaling can promote fibrosis in the tumor micro-environment. Autophagy defects in mice with residual p53 function can result in acinar to ductal metaplasia, but not in progression to carcinoma. In contrast, animals with homozygous p53 deletions show accelerated carcinogenesis when autophagy is disrupted [17,20]. Thus, the loss of function of p53 is critical for the pro-carcinogenic effect of both inflammation and autophagy.
Obesity is also associated with metabolic dysregulation, metabolic syndrome, diabetes mellitus, and hyperinsulinemia [21]. Among factors of the constellation of metabolic syndrome, hyperinsulinemia is a critical factor for carcinogenesis [21]. It can promote pancreatic carcinomas in situ in a mouse model of KRAS mutant animals [22]. Animals with high insulin levels show more extensive and higher grade PanIN, an effect that is independent of glucose levels. Hyperinsulinemia is also associated with increases of desmoplastic stroma and fibrosis in the microenvironment of PanIN. Fibrosis is a characteristic of pancreatic cancers that develop from these lesions. However, progression to pancreatic cancer was rare in animals with intact p53 even after follow-up for more than one year, confirming the protective effect of p53 against cancer progression in hyperinsulinemic conditions (Fig. 2).
Experimental data have suggested that preneoplastic pancreatic lesions in the form of acinar-ductal metaplasia can develop multifocally because underlying insulting factors can promote field cancerization [23]. However, during the transition to in situ carcinoma, the lesion becomes monoclonal due to the presence of a dominant clone that overshadows other pre-neoplastic clones. Given that mutations in TP53 appear in this stage of pancreatic cancer, the sequence of events suggests that p53 inactivation represents a usual bottleneck in the process of pancreatic carcinogenesis. Thus, TP53 mutations may be the molecular underpinning of inflammatory and metabolic causative factors of pancreatic neoplasia possibly assisted by smoking, which appears to increase the development of random additional mutations [13]. Corroborating this assumption, p53 is involved in safeguarding against the EMT (epithelial to mesenchymal transition) process, a key program in cancer cells’ acquisition of motility and invasive potential, innate in infiltrating carcinomas as described in a subsequent section.
The landscape of established pancreatic cancers
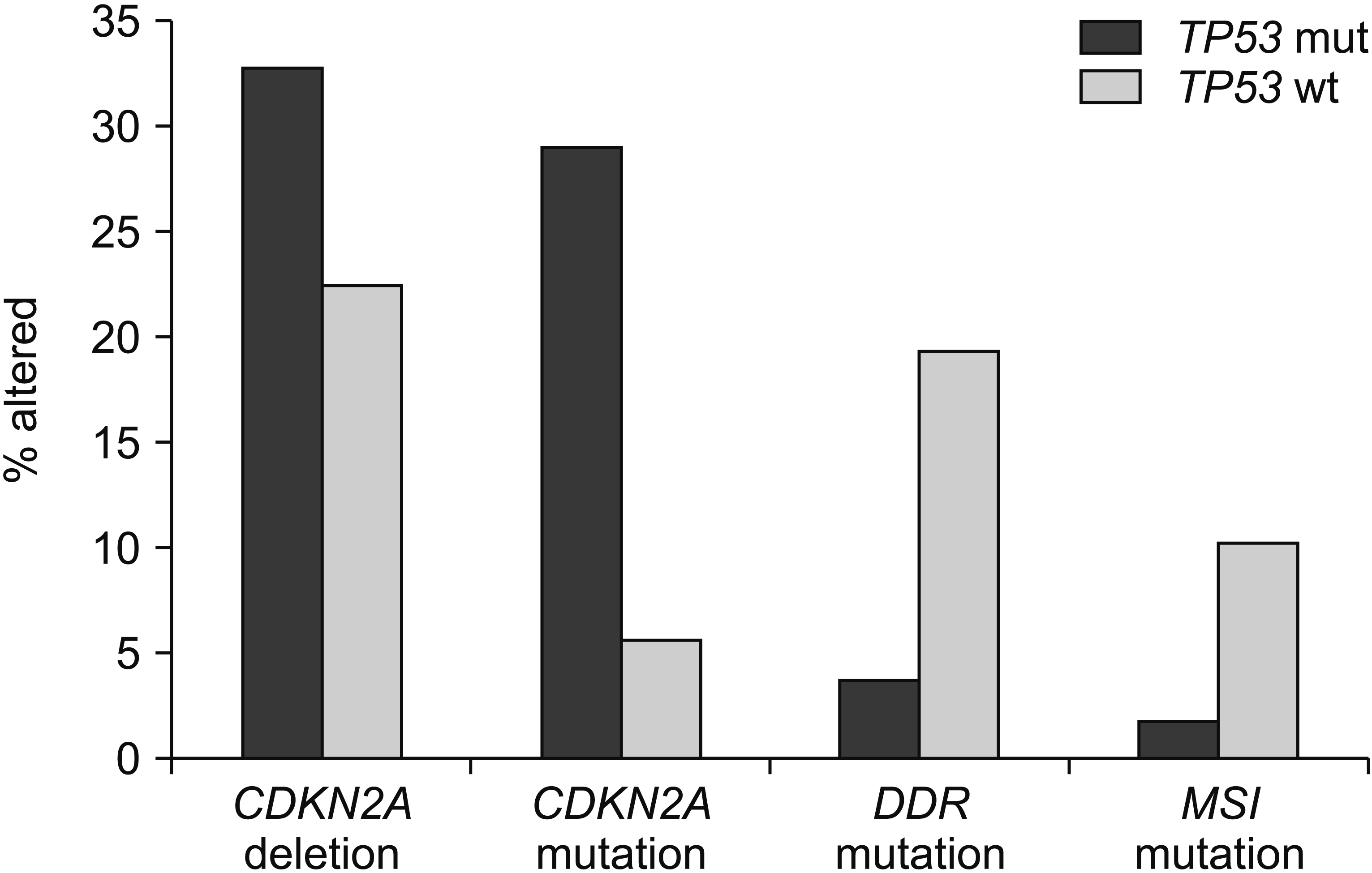
Data from published genomic series with clinicopathologic information such as TCGA, the MSK-IMPACT study, and the project GENIE are informative regarding the landscape of TP53 mutations in pancreatic adenocarcinomas [3-5]. Cases with TP53 mutations have more common underlying mutations of KRAS than cases with wild-type TP53, suggesting that KRAS mutations might develop early in favor of pancreatic carcinogenesis and build on TP53 mutations for neoplastic progression. In contrast, the smaller subset of KRAS independent cancers might rely more commonly on other molecular lesions rather than on TP53 mutations to debilitate p53 function or employ alternative pathways to obtain functional endpoints provided by p53 neutralization. Interference with p53 function is effectuated through CDKN2A homodeletion, encoding p16 and p14ARF in a sub-set of about 22% of TP53 wild-type pancreatic cancers. Homodeletion of CDKN2A is even more common in TP53 mutant cancers. It is observed in one-third of cases (Fig. 2). These data imply that deletion of p14ARF alone is insufficient to neutralize the function of intact p53. However, it provides additional advantage to cancers with a mutant p53. A putative mechanism of interference of deletions of p14ARF in cases with heterozygous p53 mutations involves a deeper suppression of the ratio of wild-type p53 to mutant p53, given that the wild-type p53 is the preferred client of MDM2 ubiquitination. In addition, p14ARF has tumor suppressing actions independent of p53 in pancreatic cancer [24]. Deletions of the In CDKN2A locus can lead to loss of the overlapping locus encoding for p16, a cell cycle inhibitor, providing additional advantage for cancer cells by neutralizing normal cell cycle control of the cyclin D/CDK4 or CDK6/Rb pathway and by instigating proliferation. Similar to deletions in the locus, mutations of CDKN2A that do not affect the availability or function of p14ARF, but still neutralize p16, are more prevalent in TP53 mutant pancreatic cancers (Fig. 3). This suggests that cell cycle dysregulation is of particular benefit in the molecular environment with absent or decreased p53 function, where cells have already lost cell cycle breakpoint provided by a functional p53.
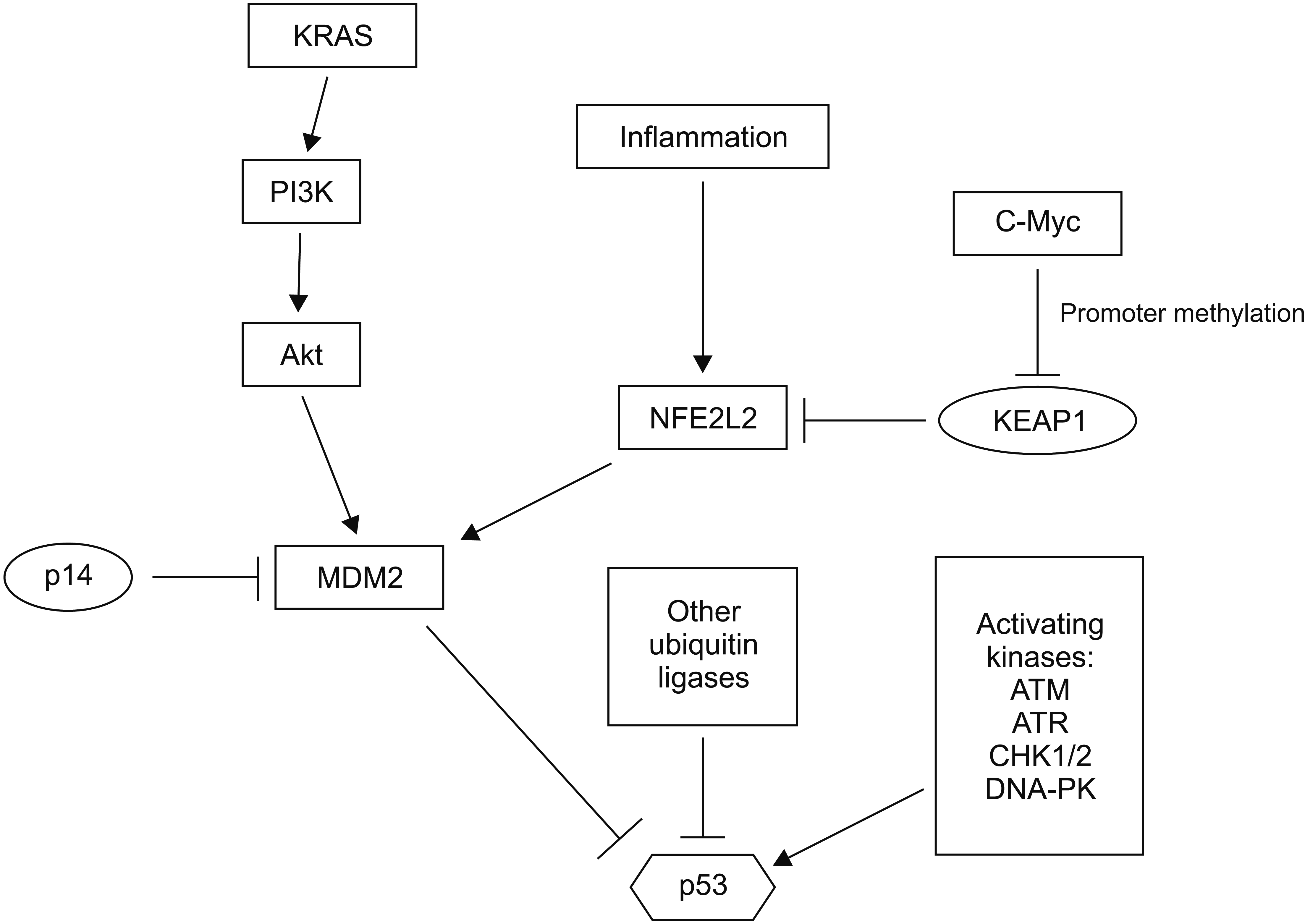
Other mechanisms of p53 functional inactivation in the absence of or in addition to TP53 mutations are often present in pancreatic carcinomas (Fig. 4). Transcription factor NFE2L2 (also called NRF2) is a master regulator of cellular detoxification response through transcription of enzymes required for NADPH production and glutathione reduction [25]. NFE2L2 is also a master regulator of normal proteostasis by regulating the transcription of proteasome structural genes [26]. NFE2L2 is a target of the activated KRAS cascade. It is upregulated in pancreatic carcinogenesis, even in pre-invasive phases [27]. Chronic inflammation further upregulates NFE2L2 by decreasing its interaction with a negative regulator KEAP1, leading to a decreased degradation by the proteasome [28]. Promoter methylation may also suppress KEAP1 transcription, a process that is facilitated by oncogene c-Myc [29]. MDM2 is a target of NFE2L2’s transcriptional activity. Thus, upregulation of this master transcription factor will result in an increased destruction of p53 [28]. In addition, MDM2 can reduce p53 activation through ubiquitination of kinase GSK3, an activator of acetyltransferase Tip60 that can acetylate and activate p53 [30]. Acetylation of p53 at lysine 120 by Tip60 favors the apoptotic program of p53 over cell cycle arrest [31]. Other targets of MDM2 such as Notch1 that are upregulated could further promote pancreatic carcinogenesis. This mechanism explains how chronic inflammation (due to chronic pancreatitis, alcohol use, or associated with obesity) co-operates with KRAS mutations to neutralize p53 and promote pancreatic carcinogenesis, even before TP53 mutations occur.
Other putative genetic lesions that may debilitate p53 signaling include mutations in kinases that activate p53. Mutations in kinases ATM and ATR have been observed in 4.5% and 2.2% of pancreatic cancers in TCGA cohort and in 2.9% and 0.3 % of pancreatic cancer cases in the MSK-IMPACT cohort, respectively [3,5]. Besides ATM and ATR, three different key p53 activating kinases, CHK1, CHK2, and DNA-PK encoded by gene PRKDC, are mutated in isolated cases. In addition to MDM2 that is regulated by p14ARF, several other ubiquitin ligases, including PIRH2 (p53 Induced RING H2 protein) and COP1 (Constitutive Photomorphogenic 1) RING finger type ligases (similar to MDM2) and HECT type ligase HUWE1 (HECT, UBA and WWE containing E3 ubiquitin ligase 1 also known as ARF-BP1 or MULE), also contribute to the tight physiologic regulation of p53 [32]. Increased activities of these alternative ligases could neutralize p53 function. However, each of them has several additional client proteins, including critical oncogenes. Down-regulation of such critical oncogenes would be deleterious for cancer cells. It could counter-select lesions in these ligases. As an example, HUWE1 can ubiquitinate and promote the degradation of oncogene c-Myc and its partner Miz1 and act as a tumor suppressor gene, at least in some cellular contexts [33]. Another target of HUWE1 for proteasomal degradation is the p53 activating, DNA damage response kinase CHK1 [34]. E2 conjugating enzymes as proteins that transfer target proteins to E3 ubiquitin ligases for ubiquitination also play a role in outcomes of ubiquitination. In the case of p53, E2 conjugating enzymes may influence protein degradation or stabilization in an inactive state, which can be reversible [35].
The cooperation between KRAS activation and p53 neutralization is a cornerstone of human pancreatic carcinogenesis. Additional oncogenes have been identified in mouse models of pancreatic carcinogenesis, including Myc, Yap1, and Nfkb2 [36]. The dosage of mutant Kras also appears to be important in the progression of pancreatic cances. In human pancreatic cancer, c-Myc is amplified in 12.6% of cases in TCGA. C-Myc amplifications are seen almost exclusively in TP53 mutated cancers.
p53 and autophagy in precancerous lesions and invasive cancers
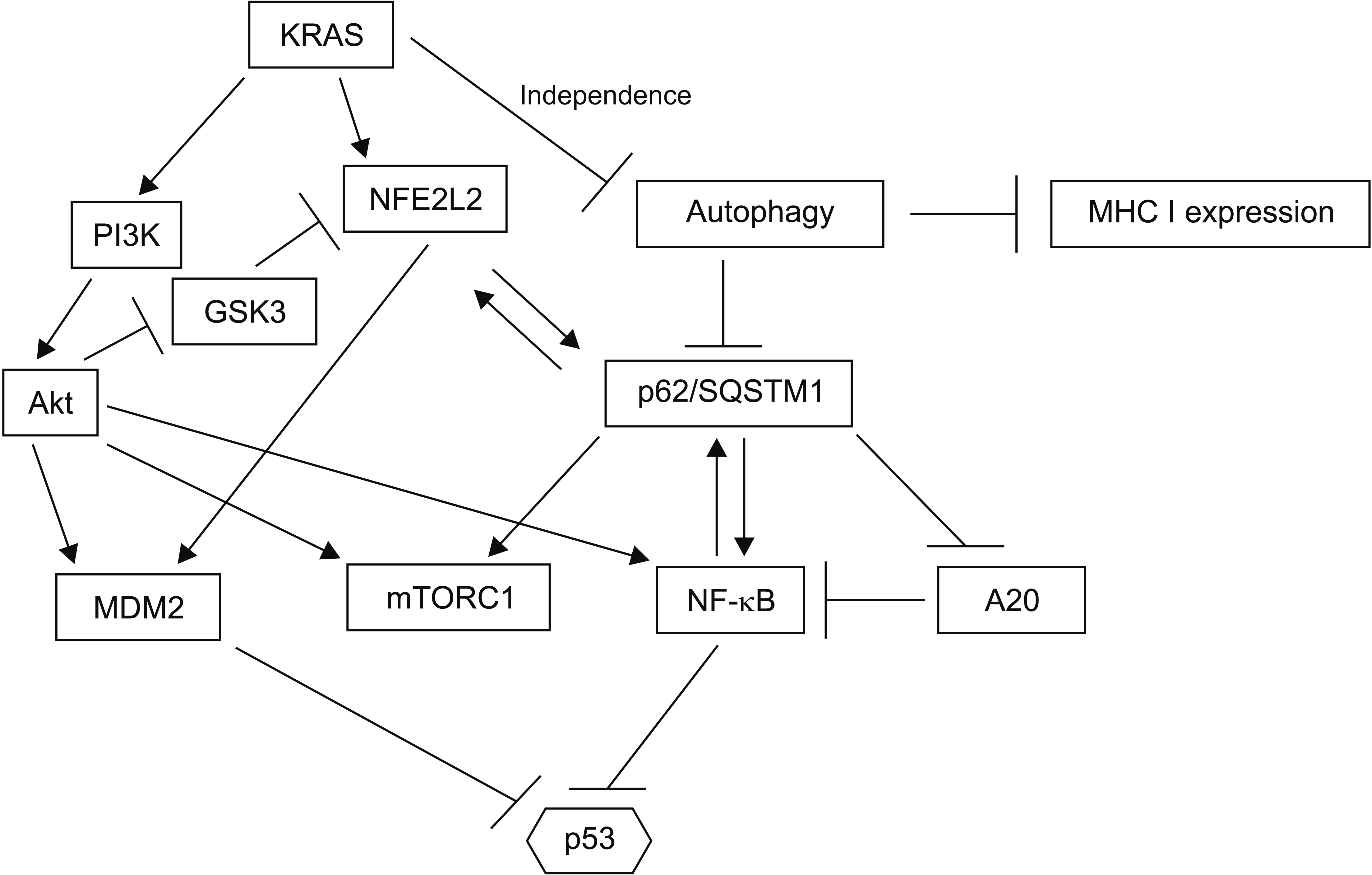
Autophagy is a physiologic process for digesting cellular components under conditions of cell stress or nutrient deprivation that can lead to their destruction for use as fuels or recycling for rebuilding organelles required for sustained cellular functions [37]. In addition, autophagy provides cells with an invaluable mechanism for handling environmental stresses (for example, exposure to reactive oxygen species) that can lead to damaged proteins and organelles. On the other hand, in established cancers, autophagy may protect cancer cells from toxins (such as those used in chemotherapy), leading to chemoresistance [38]. Although roles of autophagy in pancreatic cancer remain unclear, it appears that autophagy is highly dependent on the stage of carcinogenesis. In established pancreatic cancers that are mostly defective in p53 function, autophagy plays a cancer promoting role [17]. Autophagy may also suppress the immunogenicity of pancreatic cancer cells by selectively downregulating MHC-I complexes and antigen presentation (Fig. 5) [39]. Early pancreatic neoplastic lesions that commonly have an activated KRAS without p53 loss of function can tolerate decreased autophagy, while KRAS/RAF/MAPK signaling cascade inhibition will increase the reliance of cancer cells on autophagy [40,41]. In addition, KRAS signaling can up-regulate NFE2L2 through transcriptional upregulation of the NFE2L2 gene [28].
p62/ SQSTM1 is a multidomain protein that functions as a receptor of substates destined for autophagic degradation. It is also a positive regulator of kinase mTOR and NF-κB (a transcription factor) signaling (Fig. 4) [42]. Thus, p62/SQSTM1 not only has functions in autophagy, but also has functions in other cellular processes that are independent of autophagy. Impaired autophagy resulting from chronic inflammation can lead to KEAP1 sequestration by p62/SQSTM1 and NFE2L2 stabilization as described above, leading to p53 neutralization through MDM2 induction. This mechanism connects inflammation with pancreatic cancer progression. It might play a role in pancreatic cancers with p53 mutations by interfering with the residual activity of the remaining wild-type allele and in pancreatic cancers without p53 mutations.
p62/SQSTM1 may further promote pancreatic carcinogenesis by activating the NF-κB pathway, a key pathway for cell survival. The NF-κB pathway might also be cooperatively activated through KRAS activation [43]. The mechanism of NF-κB activation by p62/SQSTM1 involves inhibition of deubiquitinase A20, a negative regulator of NF-κB [44]. On the other hand, KRAS transduction through the PI3K/Akt pathway can phosphorylate kinase IKKα of the IKK complex, which then phosphorylates I-κBα (an inhibitor of NF-κB) promotes its degradation, allowing NF-κB family dimers to activate transcription [8]. Another common target of activation by p62/ SQSTM1 and KRAS is kinase mTOR as part of the mTORC1 complex [44]. mTORC1 can sense the nutrient status of cells and induce autophagy when nutrient supply is decreased. It is dysregulated by aberrant input from oncogenic KRAS which functions autonomously. Feed-forward loops exist among p62/SQSTM1, NFE2L2, and NF-κB given that the promoter of the SQSTM1 gene possesses binding sequences for both transcription factors (NFE2L2 and NF-κB) that can positively regulate the SQSTM1 gene (Fig. 4). Cooperation between p62/SQSTM1 and KRAS can neutralize p53 function through NFE2L2 and Akt mediated stabilization of MDM2. Additionally, Akt activity ensures NFE2L2 availability by neutralizing kinase GSK3, a negative regulator of NFE2L2 [45]. Thus, multiple pathways are involved in autophagy. KRAS signaling in pancreatic carcinogenesis can promote p53 neutralization, which might be subsequently reinforced by TP53 mutations.
Mutations in core autophagy genes (such as beclin 1, ATG7, ATG101, ULK1), SQSTM1, and KEAP1 occur only in isolated cases of pancreatic cancer, consistent with the dual role of autophagy in this disease [3,4]. The same dual role is found for other main functions of p62/SQSTM1, including the transcription of detoxifying enzymes. Detoxification of innate reactive species and external toxins can protect against cancer development by preventing genotoxic cellular damage caused by reactive species and toxins. However, in established cancers, detoxifying enzymes can protect cancer cells from damage caused by chemotherapy drugs, leading to chemoresistance [26].
MUTATIONS OF p53 IN PANCREATIC CARCINOGENESIS: THE ROLE OF EPITHELIAL TO MESENCHYMAL TRANSITION AND PLURIPOTENCY IN PANCREATIC CANCER DISSEMINATION
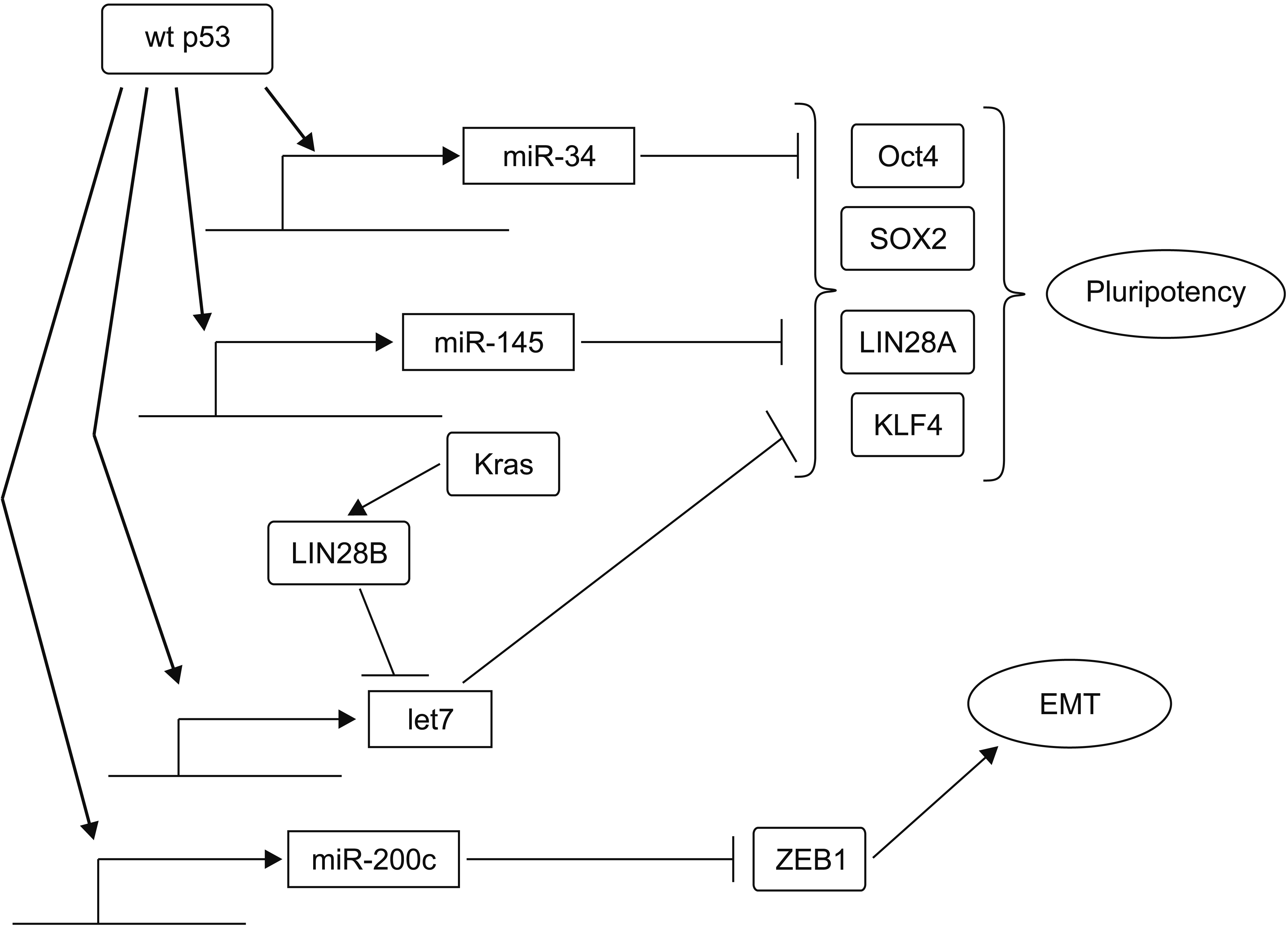
EMT is a physiologic process that is important for development and wound healing. However, it is exploited by cancer cells to promote their motility and metastasis [46]. The network of EMT encompasses a panel of core transcription regulators of ZEB and Snail families that can suppress epithelial adhesion molecules and epithelial cell polarization[30]. Loss of p53 function in pancreatic cancer cells can promote EMT by upregulating ZEB1, a zinc-finger transcription factor [47]. The mechanism, as identified in other cancers, involves stimulation of the transcription of micro-RNA miR-200c, a suppressor of ZEB1, by p53, which is lost when p53 function is absent due to its mutations or protein loss (Fig. 6) [48]. Thus, loss of function of p53 in later stages of pancreatic cancer development provides programs necessary for cell motility and metastasis that are absent in pre-invasive phases of the disease. EMT is a dynamic process that is not complete in cancer, with some features of epithelial cells retained. EMT of cancer allows cells to revert to an epithelial phenotype by reacquiring epithelial features in a process called Mesenchymal to Epithelial Transition (MET). These two transitions (EMT and MET) are collectively referred to as epithelial to mesenchymal plasticity. Related to this plasticity is the developmentally inherent phenotype of stemness, which is an embryonic development feature suppressed in adult tissue cells but reactivated in cancer cells. P53 has intimate connections with stemness programs, blocking adult differentiated cells from dedifferentiation by interfering with core stemness networks [49]. In pancreatic cancer cells with p53 mutations, micro-RNA miR-34, a p53 transcription target, is downregulated. Its restoration by transfection decreases a subset of cells with tumor-initiating capacity (Fig. 6) [50]. P53 can also activate miR-145. In cooperation with miR-34, p53 targets transcription factors of the pluripotency network such as Oct4, KLF4, LIN28A, and SOX2 [51]. Another micro-RNA, let7, might be a p53 target after its activation through ATM kinase signaling, at least in some types of cancer [52]. The let7 micro-RNA family is a barrier to stemness. It is downregulated in pancreatic cancer cells though the activity of KRAS, which activates LIN28B, an inhibitor of let7 [53]. Thus, these two programs of EMT and stemness are intertwined to facilitate cancer development and progression. p53 provides protection against untimely activation of both programs in adult tissues [54]. EMT and stemness in pancreatic epithelial cells are associated with loss of epithelial cell polarity and epithelial identity in acinar cells of pancreatic ducts. Acinar cells can lose their normal profile and transform to a ductal cell phenotype with an eventual progression towards loss of duct formation capability in later stages of carcinogenesis [28].
Mutations in epigenetic modifiers such as the SWI/SNF component ARID1A play a role in promoting the mesenchymal phenotype in early stages of pancreatic carcinogenesis [55]. Interestingly, mice with loss of ARID1A in a background of KRAS mutations developed lesions with IPMN morphology rather than PanIN morphology. Mice with both KRAS and TP53 lesions also developed neoplasms that were more cystic than animals with isolated KRAS activating mutations or p53 knock-down. In both scenarios, ARID1A loss enabled cells to acquire mesenchymal features. In addition, proteins of the EMT program such as Snail1 and PDGFRβ were upregulated [55]. ARID1A is important for the expression of acinar cell specification factors PDX1 and MIST1, suggesting that the EMT program is embedded in the acinar-ductal metaplasia program that underlies the initial phase of pancreatic carcinogenesis [56,57].
Development of EMT is associated with cancer therapy resistance, a phenomenon observed in pancreatic cancer and cells with DNA Damage Response (DDR) defects due to mutations in kinase ATM [58]. These cells are sensitive to PARP inhibitors because they are deficient in homologous recombination. However, they can develop drug resistance by undergoing EMT and up-regulating multidrug resistance transporters. Loss of function of p53 due to mutations in TP53 could act as an additional player in drug resistance both by promoting EMT and interfering with the sensing of DNA damage downstream of kinases ATR and DNA-PK. Kinases ATR and DNA-PK may still be functional in ATM mutant cells. Failure to induce micro-RNA let-7 downstream of the ATM-p53 axis, as described above, may contribute to EMT and resistance development [52].
Mutations in EMT and pluripotency core network genes are rarely observed in pancreatic cancers. In the TCGA pancreatic cancer cohort of 179 cases, mutations of ZEB1, ZEB2, KLF4, SOX2, and LIN28A with unknown oncogenic significance were observed in 1 to 3 cases each. No cases with mutated SNAI1, SNAI2 (encoding for Snail1 and Snail2, also called Slug), or OCT4 were present [3]. This suggests that the optimal dose and function of these core regulators need to be maintained in pancreatic cancer cells. More subtle perturbations through post-transcriptional regulations, which may be reversible, rather than permanent changes of gene dosage and function, might be more beneficial for cancer cells. However, c-Myc, another oncogene with close ties to pluripotency and reprogramming, is amplified in 12.6% of pancreatic cancers and other cancers [3]. Reprogramming and stemness in KRAS mutant cells are promoted by c-Myc [59]. The mechanism of c-Myc in promoting stemness, in contrast with other core stemness factors, involves a permissive function of enhancers of genes that regulate stem cell identity [60].
PANCREATIC CANCERS WITHOUT KRAS OR TP53 MUTATIONS
TP53 mutations are clearly the preferred way used by cancers to inactivate the function of p53 protein, as denoted by much higher rates of TP53 mutations in pancreatic cancer and other cancers than individual rates of mutations in other protein regulators of p53 activity. However, the aggregate rate of molecular lesions in regulators of p53 function is significant. These lesions include mutations or copy number alterations in p53 activating kinases and regulators of ubiquitin ligases that can inhibit p53, such as p14ARF deletions. They might provide the framework of carcinogenesis in cases without TP53 mutations. Pancreatic cancers without TP53 mutations more commonly lack KRAS mutations than those with TP53 mutations. In TCGA, 84.1% of TP53 mutated pancreatic cancers possess KRAS mutations, whereas only 37.5% of TP53 wildtype pancreatic cancers possess KRAS mutations.
Cases with neither KRAS mutations nor TP53 mutations represent 27.2% (50 of 184 cases) of pancreatic cancers in TCGA and 6.25% (24 of 384 cases) of pancreatic cancer cases in the MSK-IMPACT study. The latter study had a higher KRAS mutation rate in 90% of patients. Mutations in oncogene GNAS, ATM, NF1, FBXW7, SMAD4, and several epigenetic regulators (ATRX, DNMT3A, KMT2A, KAT6A and ARID1A) have been observed in more than one case each in pancreatic cancers without KRAS or TP53 mutations in TCGA. Deletions of the 9p21.3 locus harboring p14ARF and p16 are less prevalent in pancreatic cancers without TP53 mutations than in TP53 mutated counterparts. They are observed in 10% of KRAS and TP53 wild-type pancreatic cancers. About 80% of 9p21.3-deleted cases contain additional deletions of the neighboring MTAP gene encoding for methylthioadenosine phosphorylase. This enzyme is involved in purine biosynthesis. Its absence could sensitize cells to antimetabolite drugs. Deletions of 1p42 harboring TRIM genes encoding for p53 regulators and amplifications of the 8p11.23 locus harboring several oncogene candidates observed in subsets of breast cancers, squamous lung, esophageal, and bladder carcinomas are also present in pancreatic cancers with neither KRAS mutations nor TP53 mutations [61]. The absence of KRAS and TP53 mutations, together with maintenance of SMAD4 expression, is enriched in a small series of long-term survivors of pancreatic cancer [62].
Compared with TP53 mutant pancreatic cancers, TP53 wild-type cancers display a significantly higher mutation rate for genes participating in DNA damage response (DDR) such as BRCA1, BRCA2, BRIP1, RAD51C, ATM, and CDK12 that are collectively mutated in 19.5% of TP53 wild-type cases in TCGA, but only in 3.7% of TP53 mutant pancreatic cancer cases (Fig. 2). Similarly, TP53 wild-type cancers display a significantly higher mutation rate in mismatch repair genes MSH2, MSH3, MSH6, PMS1, PMS2, and MLH1 and in proof-reading polymerases POLE and POLD1 than TP53 mutated pancreatic cancers (10.4% versus 1.9%). In contrast, mutations in CDKN2A encoding for p16 are more common in TP53 mutated cancers than in TP53 wild-type pancreatic cancers (29% versus 5.6%).
Another player in pancreatic cancer with a particular role in p53 wild-type cases is peroxisome proliferator activated receptor gamma (PPARγ), a nuclear receptor family transcription factor with a role in pancreatic cancer [63]. PPARγ phosphorylation at serine S273 and its interaction with wild-type p53 can prevent apoptosis induced by p53 activation in response to DNA damage in lung carcinoma cells [64]. Reciprocally, p53 can interfere with the transcriptional activity of PPARγ by suppressing the stability of PPARγ co-activator PGC1α [65]. In pancreatic cancer cells with KRAS mutations, Hes, a notch transcription regulator, is upregulated downstream of NF-κB. It can promote the suppression of PPARγ [66]. Thus, either PPARγ protein inhibition or transcriptional suppression of PPARγ may impair p53 activity in wild-type pancreatic cancers.
TARGETING p53: THERAPEUTIC AVENUES IN PANCREATIC CANCER
Determining the status of p53 in human pancreatic cancers has dual values. First, p53 status can serve as a biomarker of response to established chemotherapy drugs and regimens such as gemcitabine and combination chemotherapies. Second, it may inform therapeutic targeting using evolving personalized strategies.
Mutations of TP53 are associated with gemcitabine resistance in pancreatic cancer cells in vitro [67]. Treatment of these cells with a combination of gemcitabine and p53 re-activating molecules CP-31398 and RITA (Reactivator of p53 and Inducer of Tumor Cell Apoptosis) has a synergistic effect in reducing proliferation and inducing apoptosis (Table 1). RITA is also an activator of wild-type p53. Increased sensitivity to a combination of RITA with gemcitabine is observed in p53 wild-type pancreatic cancer cells [67]. Moreover, RITA can sensitize resistant colorectal cancer cells to 5-fluoro-uracil and oxaliplatin independently of their p53 mutation status [68]. These data imply that restoring the function of p53 and its activation are critical for chemosensitization of pancreatic cancer cells to a wide range of DNA damaging chemotherapeutics. Germ cell tumors as prototypical chemosensitive tumors universally possess a wild-type p53, consistent with the notion that p53 activity is a prerequisite of cancer cell sensitivity to chemotherapy [49]. However, the reverse is not always true as pancreatic cancer cells with wild-type p53 may still be resistant to oxaliplatin [69]. In addition, sensitivity to chemotherapy and PARP inhibitors of wild-type TP53 pancreatic cancers may be indirectly related to the TP53 status, but more directly related to the fact that TP53 wild-type pancreatic cancers have a higher prevalence of mutations in DDR related genes such as BRCA1 and BRCA2, as discussed in the previous section. A contradictory result has been reported from an analysis of patients in the CONKO-001 trial which has compared an adjuvant gemcitabine group with an observation group of pancreatic cancer patients [70]. Patients with wild-type TP53 were found to have a better disease-free survival (DFS) than patients with mutated p53, a result concurrent with the TCGA cohort. However, they curiously derived less benefit from adjuvant gemcitabine. The hazard ratio for DFS of the group receiving gemcitabine versus the observation group was 0.79 for patients with wildtype TP53 and 0.23 for patients with mutant TP53. The reason for this discrepancy might have stemmed from differences in the dynamics of chemotherapy benefits in the adjuvant setting where the tumor burden is microscopic and factors besides treatment that can affect the ability of cancer cells to survive and establish recurrent disease. In addition, gemcitabine is less effective than platinum compounds for DDR deficient cancers. Thus, the benefit of concomitant DDR gene mutations in TP53 wildtype cancers is less pronounced.
Besides chemotherapy sensitization and direct targeting, pancreatic cancer can be treated by targeting the p53 axis using MDM2 inhibitors. These drugs even have effects on p53 mutant tumors by interfering with additional MDM2 targets. PXN822 can inhibit MDM2 by binding to the p53 binding pocket, thus inhibiting the interaction of MDM2 with p53 [71]. As a result, levels of p53 transcription target p21 are increased in pancreatic cancer cells treated with PXN822 whereas cell proliferation is decreased (Table 1). Similar effects have been observed for nutlin 3a, another MDM2 inhibitor. In cells with mutant p53 or null for p53, nutlin 3a in combination with etoposide, an inhibitor of topoisomerase II, has synergistic effects in increasing DNA damage [71]. Nutlin 3a can specifically increase double strand DNA breaks by inhibiting the interaction of MDM2 with protein Nbs1 of the MNR complex (Mre11-Nbs1-Rad50) and by inhibiting double strand repair [72]. In addition to its direct effects on pancreatic cancer cells, the fibrotic tumor stroma microenvironment of pancreatic cancer might be modified by nutlin 3a [73]. In a mouse model of pancreatic cancer, nutlin 3a treatment can modify the transcriptome of stellate pancreatic fibroblasts through p53 activation, whereas p53 function is intact in non-neoplastic cells. Nutlin 3a can also decrease fibrosis. Decreased fibrosis in the tumor microenvironment could contribute to sensitization of cancer cells to other agents through better tumor penetration.
Therapeutic interventions in pancreatic cancer, even when they do not directly affect p53, work on a the cellular environment that commonly contains a disabled p53. For example, newly introduced inhibitors of mutant KRASG12C are effective in inhibiting TP53 mutant MIA-PaCa2 pancreatic cancer cells [74]. Inhibition of mutant KRAS in this setting can create collateral dependencies on upstream tyrosine kinase inhibition or inhibition of downstream effectors of the MAPK or PI3K-Akt pathway which could be explored therapeutically [74]. KRAS-orchestrated signal transduction cascades from surface tyrosine kinases to downstream effectors can affect the status of p53 activation. They might be influenced by multifaceted p53 actions in cell cycle arrest and apoptosis promotion. Thus, therapeutic interventions affecting KRAS cascades can be modified by cells’ p53 status as part of the intracellular milieu.
Oncogene c-Myc is amplified in pancreatic cancers with TP53 mutations. c-Myc activation and over-expression remain molecular abnormalities that could not be directly targeted. However, cancers with increased c-Myc activity display increased sensitivity to proteostasis stress. They are sensitive to proteasome inhibitors in pre-clinical models [75]. These drugs have not produced benefits in a clinical trial of metastatic pancreatic cancer enrolling unselected patients. Thus, they deserve a re-evaluation using selected molecular subsets [76].
Despite the dual role of autophagy in cancer that is dependent on the stage of carcinogenesis, therapeutic exploitation of autophagy’s role in pancreatic cancer has been explored clinically through trials using autophagy inhibitors chloroquine and hydroxychloroquine in advanced pancreatic cancer, for which autophagy has a pro-neoplastic effect [77]. These two drugs, either as a monotherapy or a combination chemotherapy, have not provided clinical benefits in the metastatic stage [78,79]. The lack of significant efficacy of autophagy inhibition might be due to pharmacodynamic factors related to failure of clinically safe levels of drugs to achieve autophagy inhibition [80]. In addition, in the context of an activated KRAS and a disabled p53 in pancreatic cancer, cells can tolerate a low level of autophagy produced by inhibitory signals from the KRAS/BRAF/MAPK cascade and reduced autophagy protein levels due to disabled p53 transcription [77]. As a result, further decrease of autophagy level that is clinically relevant might be difficult to obtain from a pharmacodynamic point of view. It may require inhibition of the KRAS/BRAF/MAPK cascade which provides a synergistic effect [40]. p14ARF can induce autophagy by interfering with the interaction of Beclin 1 with Bcl2 member Bcl-xL, which inhibits Beclin 1 induction of autophagy [81]. Thus, loss of p14ARF in pancreatic cancer has a negative effect on autophagy induction. It could increase the sensitivity of these cancers to autophagy inhibition. Confirmation of loss of p14ARF as a marker of autophagy inhibition sensitivity and identification of other biomarkers associated with autophagy inhibition dependence may potentially facilitate the development of autophagy therapeutics. The future of such therapeutics in the clinic is envisioned to be in combination with other targeted drugs such as KRAS cascade inhibitors for well-characterized molecular sub-sets of pancreatic cancer with autophagy dependence.
EMT is important from a therapeutic perspective as it is associated with therapy resistance as discussed above [52,58]. The observed resistance of DDR deficient cells to PARP inhibitors through the induction of EMT invites interventions to prevent or reverse the resistance process as a means of maintaining therapeutic benefits of PARP inhibitors. Restoration of p53 activity (a key guardian against EMT) with direct modulators of mutant protein or with MDM2 inhibitors in TP53 wildtype pancreatic cancers could be an avenue to explore. Given that cancers with DDR defects commonly have wild-type TP53, combining PARP inhibitors with MDM2 inhibitors might be a viable option for drug development. A synergistic action of this combination may be reinforced by a suppressing effect of the MDM2 inhibitor nutlin-3a on the PARP enzyme in a p53-dependent manner [82]. PARP downregulation is dependent on PAR-ylation and proteasome degradation [83]. The combination of PARP inhibitor rucaparib with nutlin3a or another MDM2 inhibitor RG7388 has additive or synergistic effects in an in vitro model (cell line) of ovarian cancer, a cancer with a high prevalence of TP53 mutations [84]. Such synergistic effects in cells with defective p53 imply that the effect of MDM2 inhibition on PARP enzyme suppression might be important for developing a combination of MDM2 and PARP inhibitor. In BRCA2 proficient pancreatic cancer cells with increased KRAS/ BRAF/ MEK activation, a synergistic effect of PARP inhibition using olaparib and MEK inhibition using pimasertib has been observed both in vitro and in vivo [85]. Inhibition of MEK caused downregulation of BRCA2 levels in these cells, leading to homologous recombination defects and sensitization of these cells to both PARP inhibitors and DNA damaging agents such as evofosfamide. Interestingly, BxPC3, one of cell lines used in that study, has increased MEK activity without KRAS mutations, suggesting that MEK activation, independently of mechanism that leads to this activation, can promote DNA repair in BRCA2 proficient cells [86]. BxPC3 also possesses a non-canonical TP53 mutation at codon 220 that retains its transcriptional activity. In contrast, CFPAC-1, another pancreatic cancer cell line used in these preclinical studies, has classical mutations in both KRAS and TP53, implying that synergism of MEK inhibitors with PARP inhibitors in pancreatic cancer with increased MEK activation is independent of p53 status [85].
Integrating information from molecular pathology in pancreatic cancer therapy development remains a significant challenge. The development of new therapeutics seeks to replace the one-size-fits-all approach that has dominated pancreatic cancer therapies in the past with a more tailored approach [87]. The hope is that p53 will provide an additional gateway to therapeutic approaches based on cancer vulnerabilities.
 XML Download
XML Download