

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Prader–Willi syndrome (PWS, OMIM 176270) and Angelman syndrome (AS, OMIM 105830) are caused by the loss of expression of imprinted genes at 15q11-q13 [1]. PWS results from the absence of the paternal allele of 15q11-q13, whereas AS results from the absence of the maternal allele in the same region [2]. This phenomenon is called genomic imprinting. PWS and AS occur in one in 10,000–30,000 live births [3].
Both syndromes are neurodevelopmental disorders; however, their clinical phenotypes differ [4]. PWS is characterized by neonatal hypotonia, feeding problems, failure to thrive, hypogonadism, and childhood-onset obesity [5]. AS patients present with seizures, microcephaly, and severe developmental delay [6]. When a neonate shows neonatal hypotonia or developmental delay, PWS or AS should be considered as a part of the differential diagnosis.
Several molecular mechanisms lead to PWS and AS: deletion, uniparental disomy (UPD), imprinting defect (ID), and balanced translocation [7]. Deletion of 15q11-q13 accounts for approximately 70% of cases and is the leading cause of both syndromes. Deletions are subdivided into typical type I or II deletion, which respectively range from breakpoint (BP)1 to BP3 or from BP2 to BP3, and atypical deletion [8]. UPD is mostly due to maternal meiotic non-disjunction and accounts for 3%–30% of cases, whereas ID causes 1%–5% [9]. Loss of UBE3A function causes AS in 10%–20% of patients [10].
Because the molecular mechanisms of PWS and AS determine the recurrence risk, prognosis, and clinical phenotypes, understanding the genetic profiles of these diseases can help clinicians make an accurate diagnosis and counsel patients and their families appropriately [11]. Methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) is a diagnostic method for the simultaneous detection of copy number abnormalities and methylation status [12]. It can distinguish deletional types from non-deletional types of PWS and AS. We evaluated the clinical utility of MS-MLPA in comparison with that of methylation-specific (MS)-PCR in Korean PWS and AS patients. In addition, we investigated the relationship between clinical phenotypes and molecular mechanisms determined by MS-MLPA.
Go to : 
METHODS
Patients and samples
We retrospectively reviewed patients who underwent MS-PCR for PWS and AS in the Seoul National University Hospital (SNUH), Seoul, Korea between March 2007 and July 2018. We selected 45 PWS and 24 AS patients who provided informed consent for secondary utilization and also selected 24 patients who showed negative MS-PCR results and normal karyotypes. The medical records, including diagnosis, chief complaints, laboratory results, and other clinical information, were reviewed retrospectively. For PWS, Holm diagnostic criteria were calculated (Table 1) [5]. AS 1995 diagnostic criteria were applied for the diagnosis of AS (Table 2) [6]. This study was performed in accordance with the Declaration of Helsinki and was approved by the IRB of SNUH (IRB approval number 1811-075-985).
Table 1
Frequency of PWS Holm diagnostic criteria in 45 PWS patients
Adopted from “Prader–Willi syndrome: consensus diagnostic criteria,” by Holm VA, et al. 1993 [5].
![]()
Table 2
Frequency of AS diagnostic criteria in 24 AS patients
Adopted from “Angelman syndrome: consensus for diagnostic criteria,” Williams CA, et al. 1995 [6].
![]()
MS-MLPA
MS-MLPA was performed using archived genomic DNA and the standard protocol of the SALSA MLPA Probemix ME028-C1 PWS/AS kit according to the manufacturer’s guideline (MRC-Holland, Amsterdam, Netherlands). In brief, 200 ng of genomic DNA was denatured at 98°C for five minutes and hybridized with ME028 probe mix at 60°C for 16 hours. The product was aliquoted into two tubes: one for copy number analysis and one for methylation analysis using methylation-sensitive endonuclease. The PCR products were analyzed using an ABI 3130xl capillary sequencer (Applied Biosystems, Foster City, CA, USA) and the data were analyzed using GeneMarker v.1.51 (SoftGenetics, State College, PA, USA). To normalize peak intensities, we used internal control probe normalization, and the intensity ratios of identical probes from the sample were compared with controls.
Statistical analysis
The concordance rate between MS-PCR and MS-MLPA was calculated using Cohen’s kappa coefficient. Continuous variables were compared using Student’s t-test and Mann–Whitney U-test. All statistical tests were two-tailed and performed using SPSS version 25.0 (IBM, Armonk, NY, USA). Results were considered statistically significant at P<0.05.
Go to :
RESULTS
Comparison of MS-PCR and MS-MLPA
There were no discordant results between MS-PCR and MS-MLPA, with 45 patients diagnosed as having PWS, 24 patients as having AS, and 24 non-PWS/AS controls. Therefore, the concordance rate was 100%, and Cohen’s kappa was 1.0, which indicates perfect agreement.
Genetic subtypes of PWS and AS
Unlike MS-PCR, MS-MLPA could discriminate between the deletion and non-deletion types (Figs. 1 and 2). Among the 45 PWS patients, 26 (57.8%) had deletions on the q arm of chromosome 15: eight had a type I deletion, 17 had a type II deletion, and one had an atypical deletion (Fig. 3A). The atypical deletion ranged from SNRPN to GABRB3, which is shorter than typical types. Nineteen PWS patients (42.2%) had UPD/ID. Among the 24 AS patients, 16 (66.7%) had deletions, seven (29.2%) had UPD/ID, and one (4.2%) showed microdeletion of the AS-shortest region of deletion overlap, which corresponds to an imprinting center (IC) deletion (Fig. 2C). Of the 16 patients with deletions, seven had type I deletions, and nine had type II deletions (Fig. 3B).
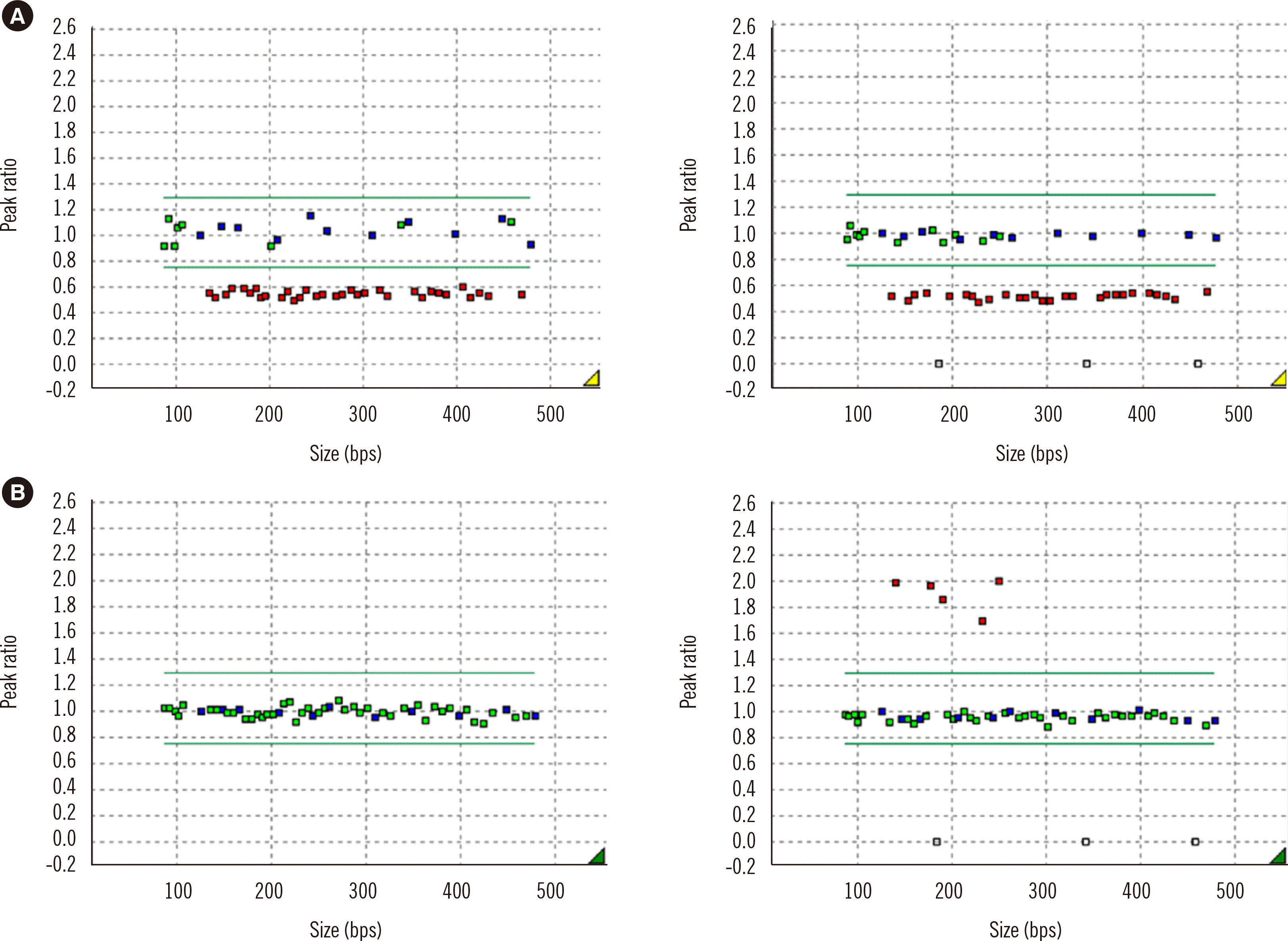 | Fig. 1MS-MLPA results of PWS patients. (A) Deletion type of PWS. (B) Non-deletion type of PWS. (Left panels: undigested, right panels: digested).
Abbreviations: MS-MLPA, methylation-specific multiplex ligation-dependent probe amplification; PWS, Prader–Willi syndrome.
|
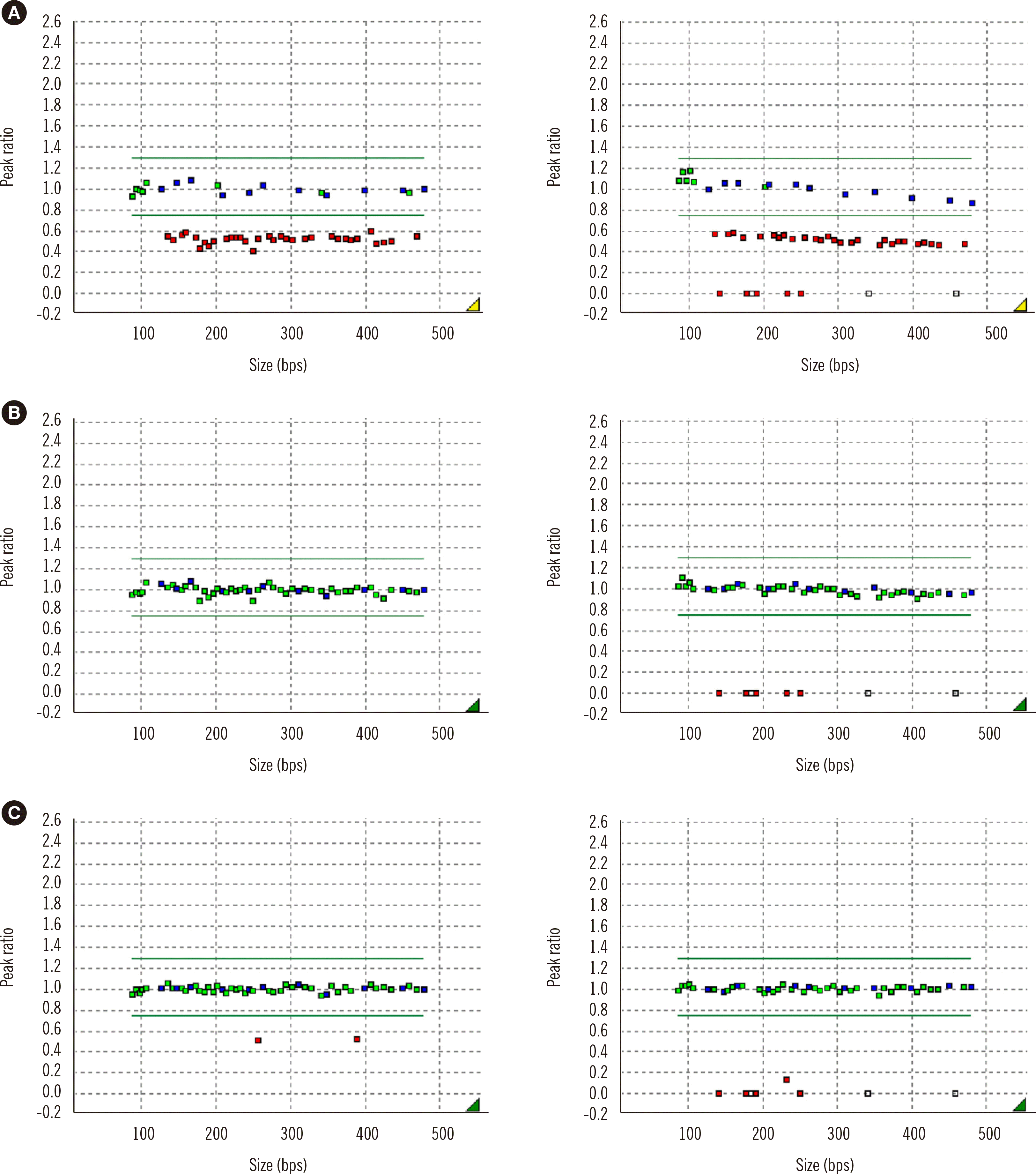 | Fig. 2MS-MLPA results of AS patients. (A) Deletion type of AS. (B) Non-deletion type of AS. (C) IC deletion of AS. (Left panels: undigested, right panels: digested).
Abbreviations: AS, Angelman syndrome; IC, imprinting center; MS-MLPA, methylation-specific multiplex ligation-dependent probe amplification.
|
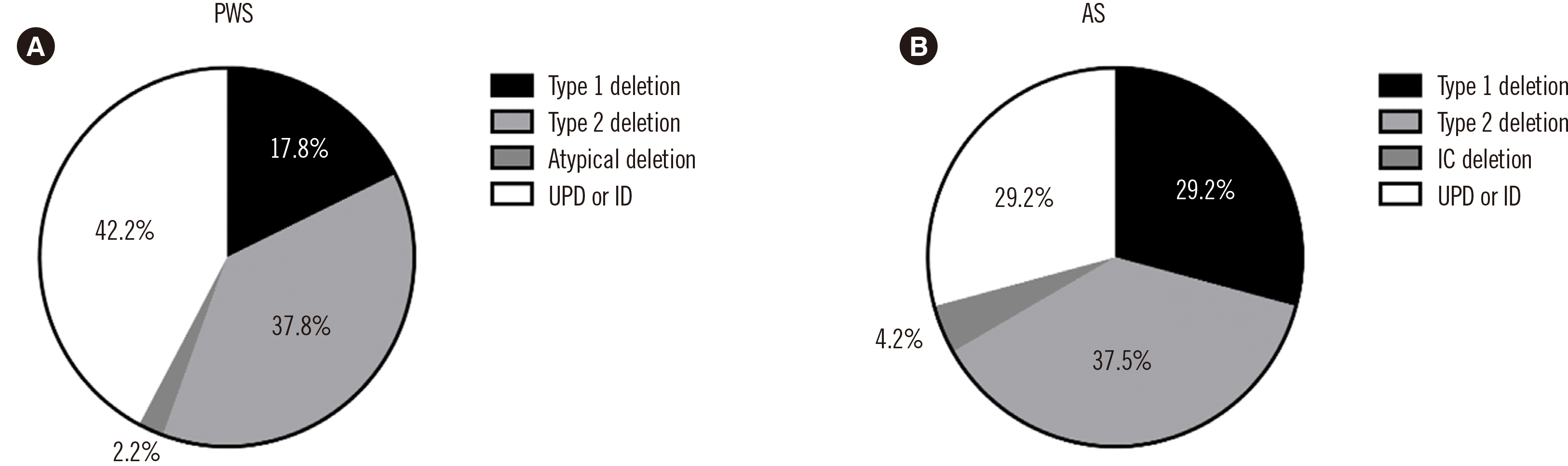
Clinical characteristics and molecular mechanisms of PWS and AS
The male:female ratio of PWS was 1.2:1, and that of AS was 0.6:1 (Table 3). The median age of PWS patients at diagnosis was four months (1–187 months), and that of AS patients was 24.5 months (9–95 months). In AS, the age at diagnosis differed according to the molecular mechanism: patients with the deletion type were diagnosed earlier than those with UPD/ID types (23.8 months vs. 55.7 months, P=0.002). Birth weight also significantly differed according to the molecular mechanism: patients with the deletion type weighed more at birth than those with the UPD type in PWS and vice versa in AS (2.7 kg vs. 2.4 kg, P=0.039 and 2.9 kg vs. 3.5 kg, P=0.035).
Table 3
Clinical features and molecular mechanisms in PWS and AS patients
![]()
PWS patients mainly showed neonatal hypotonia, developmental delay, altered mentality, failure to thrive, waddling gait, or obesity. Most AS patients (91.7%) visited our hospital for developmental delay, except two patients who had seizures and torticollis as the main problem. In PWS cases, the mean Holm score was 4.46, which is below the diagnostic criteria (Holm score 6). We observed no notable difference in the Holm scores of PWS according to the molecular mechanism or deletion range.
Go to :
DISCUSSION
Chromosomal microarray (CMA) is currently considered the first-tier diagnostic genetic test for neurodevelopmental disorders [14, 15]. However, there remains a necessity for methylation analysis, especially for imprinting disorders, because CMA is not sufficient for diagnosis of these disorders [16]. We showed that MS-MLPA can not only diagnose PWS and AS, but also reveal the underlying molecular mechanisms. We also demonstrated the relationship between molecular mechanisms and clinical characteristics of Korean PWS and AS patients. Our findings support that MS-MLPA is a useful diagnostic test for PWS and AS.
We detected the deletion type in 57.8% of PWS cases, which is in contrast to results in a previous Korean study in 2004, in which deletion accounted for 80% of PWS cases [17]. In line with our results, recent studies have demonstrated that the deletion type in PWS constitutes approximately 60% of total cases [18, 19]. Butler, et al. [18] reported that in PWS, the maternal age was higher in UPD cases than in deletion cases. Although we were not able to assess maternal age for all patients, the average maternal age in Korea is increasing yearly, from 30.0 years in 2004 to 32.4 years in 2016, according to the Korea Statistics [20]. Thus, the increase in the non-deletion type may be because UPD is more prone to occur as maternal age increases.
PWS patients with type I deletions show a more severe phenotype than those with type II deletions [21]; however, there were no significant differences in the diagnostic scores of our patients according to the deletion range. The mean Holm score of total PWS was even lower than the diagnostic score. This may be explained by poor clinical evaluation or diagnosis before clinical symptoms present due to advanced molecular diagnosis.
An atypical deletion, ranging from SNRPN to GABRB3, was detected in one PWS patient (case 39). Additional CMA results confirmed that the patient had a 2.8 Mb deletion from PWRN2 to GABRB3. He visited our hospital for lymph node enlargement at 12 years of age. At that time, he had mild learning disabilities and obesity [22, 23]. His Holm score was 5, which is higher than the mean score of PWS patients. This deletion type showed that MKRN3, MAGEL2, NDN, and C15orf2 are not indispensable for PWS symptoms [24].
One AS patient with an IC deletion (case 66) visited our hospital for developmental delay at 36 months of age. He showed a facial dysmorphism, developmental delay and generalized seizures, which indicated a diagnosis of AS. Although the probability of recurrence of IC is up to 50%, which is the highest recurrence rate for PWS/AS, his younger sister, who is the second-born child, is healthy [25].
To the best of our knowledge, this study is the largest cohort covering both PWS and AS, with clinical information. A few previous studies dealt with the molecular diagnosis of these syndromes, but most of them focused only on PWS or presented no clinical findings [2, 26].
Our study has some limitations. First, there is an intrinsic limitation to MS-MLPA in that it cannot distinguish UPD from ID. To distinguish these two mechanisms, microsatellite analysis or single nucleotide variant analysis is required [27]. However, we successfully differentiated the IC deletion type, which has a 50% recurrence risk, using MS-MLPA. In addition, we could not evaluate the behavioral or psychological status of the patients due to the lack of such information in the medical records. Lastly, this was a retrospective and single-center study. Thus, a selection bias may exist, and further large-scale prospective studies for PWS and AS are needed.
AS caused by a UBE3A variant, which accounts for 20% of AS cases, cannot be diagnosed using MS-MLPA, and further diagnostic testing is needed. To date, only two patients have been diagnosed as having AS due to UBE3A pathogenic variants in our laboratory. Therefore, we propose the use of MS-MLPA as a first-line diagnostic tool for PWS or AS, in accordance with the EMQN/ACGS guidelines [28]. The next step would be UBE3A variant analysis of samples from patients who are highly suspected of having AS, as well as maternal samples.
In conclusion, MS-PCR and MS-MLPA show perfect diagnostic concordance, and MS-MLPA can substitute for MS-PCR. In addition, MS-MLPA provides more information about the molecular mechanisms underlying the diseases and may be a helpful tool for genetic counseling of families with PWS and AS. Finally, patients who are strongly suspected of having AS and show negative MS-MLPA results should undergo additional testing.
Go to :
 XML Download
XML Download