

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Gallbladder cancer (GBC) is the 20th most incident cancer and 6th most incident gastrointestinal cancer with a dismal prognosis.1 Its poor prognosis is largely attributed to delayed diagnosis because of the absence of specific signs and lack of a reliable screening test.2 Globally, GBC rates exhibit marked regional variability, and this likely resides in the differences in environmental exposure and genetic predisposition modulating carcinogenesis, such as gallstones, chronic cholecystitis (CC), obesity, environmental exposure, and specific infections.3 However, the interplay of these factors in GBC development is still poorly understood.
One of the important hypothesis for the carcinogenic mechanism of GBC is that unresolved bacterial infection leads to gallbladder (GB) carcinogenesis.4 Several reports have shown that several strains of Salmonella and Helicobacter can be colonized in the GB and are associated with increased GBC risk.56 In addition, recent studies have provided morphological evidence and elucidated the molecular mechanism for Salmonella-induced GBC or its premalignant lesions.78 CC was commonly observed in the calculous gallbladder of mice, but premalignant lesions were only associated with chronic Salmonella infection, regardless of the presence of gallstones.8 These epidemiologic and experimental studies support the role of infection in GB carcinogenesis. However, these studies are limited to a few cultivable species, and thus have a clear limitation. Despite the proximity of the GB to the large microbial reservoir in the gastrointestinal tract, little is known about the human bile microbiome.
To overcome this limitation, the use of next-generation sequencing technology has been applied to enable microbiome profiling and to investigate the role of the microbiome in cancer development, particularly in the area of gastrointestinal malignancies.9 Traditionally, the gallbladder has been considered sterile.1011 This concept is supported by several proposed mechanisms: high concentrations of bile acids in the gallbladder function as bactericides, the Kupffer cells in the liver prevent mutagens and toxins from entering the hepatobiliary system, and secretory immunoglobulin A prevents the colonization of the microbiome.412 However, there is growing evidence that the GB bile contains an autochthonous microbiota, and its disturbance or imbalance may result in GB disease, including GBC.13
In this study, we aimed to explore the microbiomes in bile obtained from patients with a normal GB, CC, and GBC using next-generation sequencing technology. Our goal was to find a possible microbial link between chronic bacterial infection of the GB and GB carcinogenesis, and correlate the microbiome patterns with clinical, radiological, and pathological findings in these GB diseases.
METHODS
Patients and surgical procedures
A total of 25 subjects (15 patients and 10 healthy controls) were enrolled (Fig. 1). All healthy controls were living-donors for liver transplantation at the Asan Seoul Medical Center and were scheduled for right hepatectomy and cholecystectomy. Donors were examined preoperatively in the outpatient clinic for preoperative evaluation: past history was taken and medical check-up was conducted for all of them, including a computed tomography (CT) scan. All of them had no history of alcohol, smoking, or biliary disease and no comorbidities, and the laboratory and CT results were unremarkable. Among the 15 patients, 10 underwent laparoscopic cholecystectomy for the treatment of CC and 5 underwent extended cholecystectomy for the treatment of GBC at the Hanyang University Hospital.
Fig. 1
Flowchart of 25 participants. 10 Healthy controls and 15 patients with gallbladder diseases were enrolled. 10 Healthy controls were living-donors for the liver transplant and underwent right hepatectomy and cholecystectomy. Among 15 patients, 10 patients with chronic cholecystitis underwent laparoscopic cholecystectomy and 5 patients with gallbladder cancer underwent extended cholecystectomy. Bile samples were collected aseptically and analyzed.
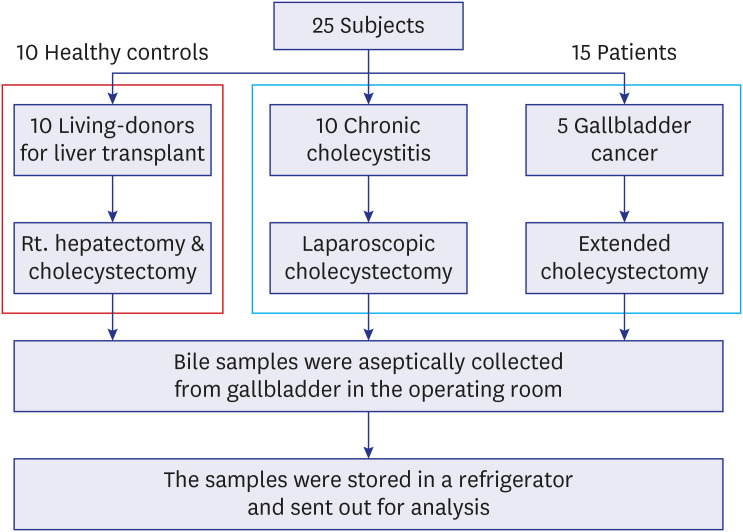
The bile of all participants were collected in the operating room from their non-perforated GBs using aseptic techniques. The samples were stored in a refrigerator and delivered to the laboratory while being maintained at a low temperature. All procedures were performed aseptically.
Preoperative radiologic evaluation
Abdominal CT examinations were performed using multi-detector CT (MDCT) machines, and a dose of 2 mL/kg of contrast agent (Ultravist 300, Schering Korea, Seoul, Korea) was administered intravenously at a rate of 4 mL/s. The MDCT scans were obtained at the arterial and portal phases at 35 s and 70 s, respectively, and transverse, coronal, and sagittal images were reconstructed at 3 mm intervals with a 3 mm section thickness.
The preoperative CT images were reviewed by an experienced board-certified reviewer who was blinded to the patients' clinical data. We analyzed the imaging features of CC and GBC used in the previous reports: the thickness of the GB wall, enhancement patterns seen on arterial and portal phase scans, pericholecystic lymph node size (larger than 1 cm in short diameter), subserosal edema, pericholecystic fat stranding, the presence of stones, the number of stones, and the size of the largest stone.141516 Additionally, we measured the volumes of the stones and GBs using Barco's Voxar 3D software. Receiver operating characteristic analysis was performed to determine whether these parameters could help classify the two groups.
Pathological examination
Cholecystectomy specimens were fixed in 10% neutral buffered formalin for more than 6 h. After a careful gross examination, the representative tumor sections and cystic duct margin were selected for microscopic evaluation. If present, the cystic duct lymph node was processed. The tissues were processed using routine methods and embedded in paraffin. Sections (4 μm in thickness) were stained with hematoxylin and eosin, and evaluated with a light microscope by expert pathologists. The pathological evaluation included tumor site, size, histological type, grade, depth of invasion, lymphovascular invasion, lymph node metastasis, and resection margins. For CC, inflammatory lesions were semi-quantitatively scored according to the modified criteria of a previously described method.17 The detailed criteria for scoring is described in Supplementary Table 1. Radiologic and pathologic findings were compared in patients with CC according to the state of dysbiosis, and positive centered log-ratio (CLR) value were considered as a dysbiotic microbiome.
Preparation of 16S rRNA and metagenomic sequences
Bacterial DNA was extracted using a Stool Power Water® DNA Isolation Kit (MO BIO, Carlsbad, CA, USA). Libraries were prepared according to the GS FLX Library Prep Guide. The 16S universal primers 27F (5′-GAGTTTGATCMTGGCTCAG-3′) and 518R (5′-WTTACCGCGGCTGCTGG-3′) were used for amplifying the 16S rRNA genes. The sequencing was performed on a Genome Sequencer FLX Plus (454 Life Sciences) by Macrogen Ltd. (Seoul, Korea).
For Illumina sequencing of the 16S rRNA genes, samples were prepared according to the Illumina 16S Metagenomic Sequencing Library protocols. The barcoded primer sequences were 341F (5′-CCTACGGGNGGCWGCAG-3′) and 806R (5′-GACTACHVGGGTATCTAATCC-3′). The paired-end (2 × 300 bp) sequencing was performed by Macrogen using the MiSeq™ platform (Illumina, San Diego, CA, USA).
Three whole metagenomic samples were prepared according to the Illumina protocols. For a 350 bp size insert, one microgram of genomic DNA was fragmented using Covaris. The final ligated product was quantified using qPCR according to the qPCR Quantification Protocol Guide and qualified using the Agilent Technologies 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA). Sequencing was performed using the HiSeq™ 2000 platform (Illumina).
Taxonomy profiling of the 16S rRNA sequences
Paired-end reads were assembled using FLASH.18 The low-quality ends (Phred quality score < 20) were trimmed and short reads were filtered out using Sickle. The operational taxonomic units (OTUs) were detected by cd-hit-est19 with 99% sequence identity. Only OTUs with sizes larger than 10 were used in the downstream analysis.
Taxa were assigned by RDP naïve Bayesian classifier20 at the family level. For genus and species level classifications, the representative sequences of OTUs were searched against the NCBI 16S ribosomal RNA database downloaded on June 12, 2020, using BLASTn (E-value < 1.0e-10 and identity > 94%). Among the families and genera assigned, the taxa were filtered out as putative contaminants according to two criteria: i) taxa distributed at a constant ratio in most of the samples of two groups21; ii) taxa constituting less than 1% of the population.22 The number of reads in each taxon was normalized using the CLR method21 implemented in the skbio v0.5.6 python package.
Analysis of bacterial composition in the microbiome using shotgun sequencing data
After removing host contaminated, low-quality (Phred quality score < 20 and length < 60% of read length), or ambiguous sequences using Sickle, the retained reads were assembled using MEGAHIT (v.1.1.3).23 The contigs over 1 kbps were translated into protein sequences using FragGeneScan (v.1.20)24 with the options of -w 1 -t complete.
For the species identification, read mapping was performed against the bacterial reference genomes using Bowtie225 with the “--sensitive-local” option. Genome coverage was calculated as the ratio of the genome sequence to the reads mapped. Read depth in a position was normalized using counts per million (CPM). The CPM value was calculated by dividing the number of mapped reads in each position by the total number of reads and then multiplying by one million.
Phylogenetic analysis
Phylogenetic trees were constructed with marker genes. For GBC1 and GBC2, RNA polymerase Rpb2 (PF00562.26) was used. A sequence search (e-value < 1E-10 and coverage > 50% of the pHMM model length) was performed using HMMscan.26 For CC4, three marker genes of ribosomal methyltransferase were used.
Multiple sequence alignment was performed using MUSCLE (v.3.8.31).27 Phylogenetic trees were constructed using MEGA6 (v.6.06).28 The phylogenetic relation was inferred using the maximum likelihood method based on the JTT matrix-based model. Initial trees for the heuristic search were obtained by using the neighbor-joining method with a matrix of pairwise distances estimated using a JTT model. The tree was drawn to scale, with branch lengths measured in the number of substitutions per site. The positions with less than 95% site coverage were eliminated.
Statistical analysis
SPSS version 18.00 (SPCC Inc., Chicago, IL, USA) was used for statistical analysis. Quantitative variables are reported as mean ± standard deviation and qualitative variables as percentages. The Mann-Whitney U test was used for comparing the two groups. P < 0.05 was considered statistically significant.
Ethics statement
This study was approved by the Hanyang University Hospital (Approval No. HYI-15-144-4) and Asan Seoul Medical Center (Approval No. 2017-0046). Written informed consent was obtained from all participants prior to enrollment in the study. All experiments were performed in accordance with the Declaration of Helsinki.
RESULTS
Summary of clinical characteristics of the study population
Table 1 summarizes the clinical characteristics of the patients enrolled. There were no significant differences between the two groups (healthy control and patients) for the following variables: the proportion of male gender, body mass index (BMI), alkaline phosphate (ALP), total bilirubin, albumin, cholesterol, and triglyceride (TG). However, aspartate aminotransferase (AST), alanine aminotransferase (ALT), prothrombin time (PT), and high-density lipoprotein cholesterol (HDL-C) showed significant differences, probably because of underlying gallbladder diseases that have adverse effects on liver function and lipid metabolism. There was no difference in comorbidities among the patient group (Supplementary Table 2).
Table 1
Summary of clinical characteristics of participants
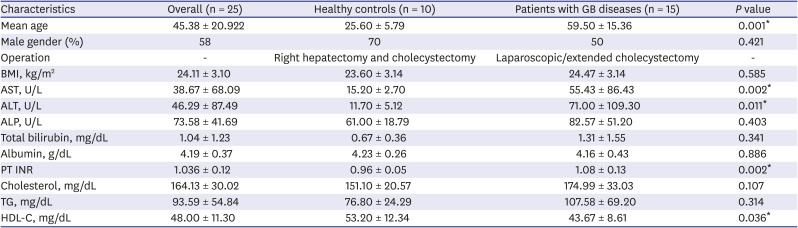
For each variable, results were expressed as mean ± standard deviation.
GB = gallbladder, BMI = body mass index, AST = aspartate transaminase, ALT = alanine transaminase, ALP = alkaline phosphatase, PT = prothrombin time, INR = international normalized ratio, TG = triglyceride, HLD-C = high-density lipoprotein cholesterol.
*Statistically significant results from Mann-Whitney U test.
Low concentration of the microbiome in normal bile
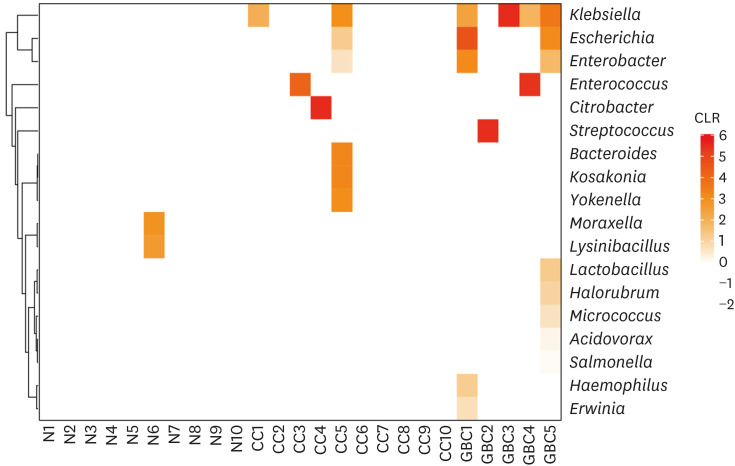
A total of 10 bile samples from healthy controls were analyzed using the 16S rRNA-based metagenomic approach. After the removal of contaminant background DNA and sequencing noise, we could not identify any meaningful signal (Fig. 2).
Fig. 2
The bacterial composition of the bile of the healthy individuals and patients with chronic cholecystitis and gallbladder cancer. The number of sequences profiled at the genus level was normalized using the CLR method. The normalized values under 0 were colored white. The maximum normalized value of pseudo-count was −0.3072.
CLR = centered log-ratio, N = normal control, CC = chronic cholecystitis, GBC = gallbladder cancer.
Citrobacter species in patients with CC
Among the 10 bile microbiomes of the CC patients, four samples showed signals for bacterial existence. The sample CC1 contained a small population of Klebsiella. Enterococcus and Citrobacter were found in CC3 and CC4, respectively: 18% in CC3 and 100% in CC4. The most diverse Enterobacteriaceae bacteria were found in CC5, such as Enterobacter, Escherichia, Klebsiella, Kosakonia, and Yokenella, in addition to Bacteroides.
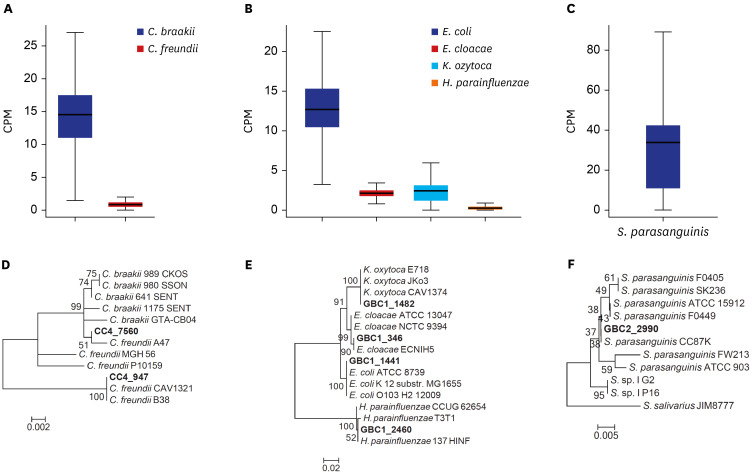
For further investigation, metagenomic shotgun sequencing was performed for CC4. Since several C. braakii and C. freundii strains have been found in the hospital environment, the genomic and phylogenetic analyses were performed with Citrobacter strains found in the CC sample (Supplementary Table 3). Genomic analysis showed that two distinctive species, C. braakii and C. freundii, exist in the bile microbiome of the CC patient. The mean CPM values for C. braakii and C. freundii were 14.52 and 0.84, respectively (Fig. 3A). Phylogenetic analysis consistently showed that C. braakii and C. freundii strains in CC4 constructed a lineage with known C. braakii and C. freundii strains including A47, MGH56, and P10159 (Fig. 3D).
Fig. 3
The bacterial species identified in patients with GBC and CC. The y-axis of each boxplot indicates CPM reads calculated from the result of read mapping in CC4 (A), GBC1 (B), and GBC2 (C). Phylogenetic tree of the bacterial species in CC4 and Citrobacter species (D), in GBC1 and four Enterobacteriaceae species (E), and in GBC2 and Streptococcus species (F). The proteins obtained from the bile microbiomes are in bold.
GBC = gallbladder cancer, CC = chronic cholecystitis, CPM = counts per million.
Enterobacteriaceae in the bile microbiomes of the cancer patients
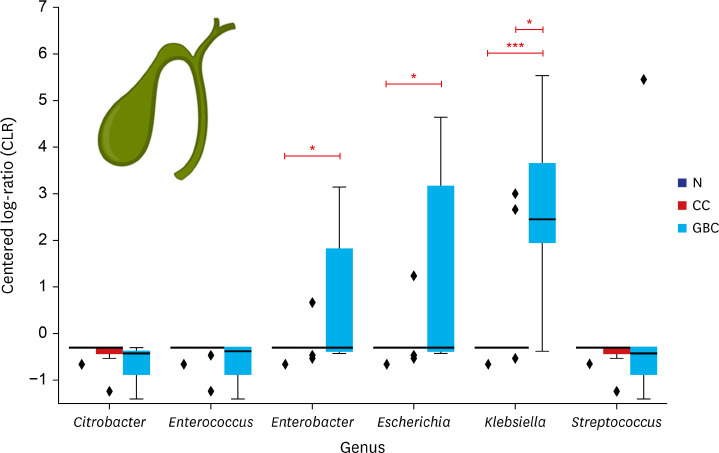
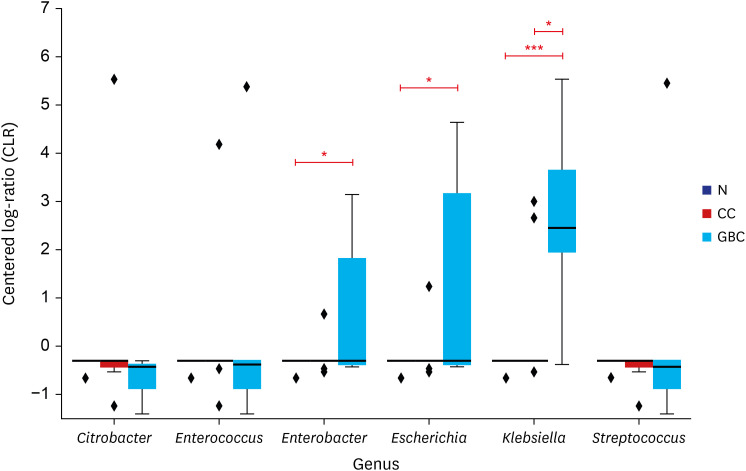
In four bile microbiomes out of five GBC patients, significant signals were detected for Enterobacteriaceae (Fig. 2). At the genus level, they showed two different patterns of bacterial compositions. In GBC1 and GBC5, multiple Enterobacteriaceae species such as Escherichia, Enterobacter, and Klebsiella were observed. In GBC3 and GBC4, Klebsiella was found. Notably, Enterococcus was the dominant genus in GBC4. Among the bacteria showing strong signals, the distributions of Enterobacter, Escherichia, and Klebsiella showed significant differences between normal people and GBC patients (P < 0.01 for Enterobacter and Escherichia; P < 0.05 for Klebsiella) (Fig. 4). Additionally, the distribution of Klebsiella showed a significant difference between CC and GBC patients (P < 0.05).
Fig. 4
The distribution of the six major bacteria in three different clinical groups.
Genera that show significant difference between two different groups are marked with asterisk: *P < 0.05, **P < 0.01, ***P < 0.001. Normalized values lower than −0.3072 are all derived from pseudo-count.
N = normal control, CC = chronic cholecystitis, GBC = gallbladder cancer.
To investigate such multiple Enterobacteriaceae species in more detail, shotgun metagenomic sequencing was performed for GBC1. The four major species in GBC1 were E. coli, E. cloacae, K. oxytoca, and H. parainfluenzae. The genomic and phylogenetic analyses showed that the mean CPM value for E. coli (= 12.71) was the highest, which was over three times higher than those for the other species (Fig. 3B). This is consistent with the result of the 16S rRNA analysis. Notably, the mean CPM value for H. parainfluenzae (= 0.25) was the lowest among the four species found in GBC1 (Fig. 3B). In a phylogenetic analysis using RNA polymerase Rpb2 as a marker gene, a close phylogenetic location was observed between the genes in the sample and for each Enterobacteriaceae species (Fig. 3E), indicating that E. coli, E. cloacae, K. oxytoca, and H. parainfluenzae species exist in GBC1.
Streptococcus in the bile microbiomes of the GBs of the cancer patients
A completely different bacterial composition was observed in GBC2, as compared to that found in other GBC patients. Nearly all sequences were classified as Streptococcus in the 16S rRNA profiling. Genomic and phylogenetic analyses using shotgun metagenomic sequencing data also showed that the mean CPM value against the S. parasanguinis ATCC 15912 genome was 33.82 in GBC2 (Fig. 3C) and the Rpb2 gene in GBC2 (GBC2_2990) was completely grouped with those from seven S. parasanguinis strains (ATCC_15912, F0449, F0405, SK236, CC87K, FW213, and ATCC_903) (Fig. 3F).
Radiologic findings of CC with or without a dysbiotic microbiome
Among 10 patients with CC and gallstone, preoperative CT was performed in seven patients. The results of the radiological findings are summarized in Supplementary Table 4. Among the various morphological CT imaging findings, the number of stones was not significant (P = 0.432), but the size of the largest stone was significantly different on comparison between the two groups (P = 0.014) (Fig. 5A and B). The group with a dysbiotic microbiome pattern displayed a tendency towards larger stone volumes (P = 0.190).
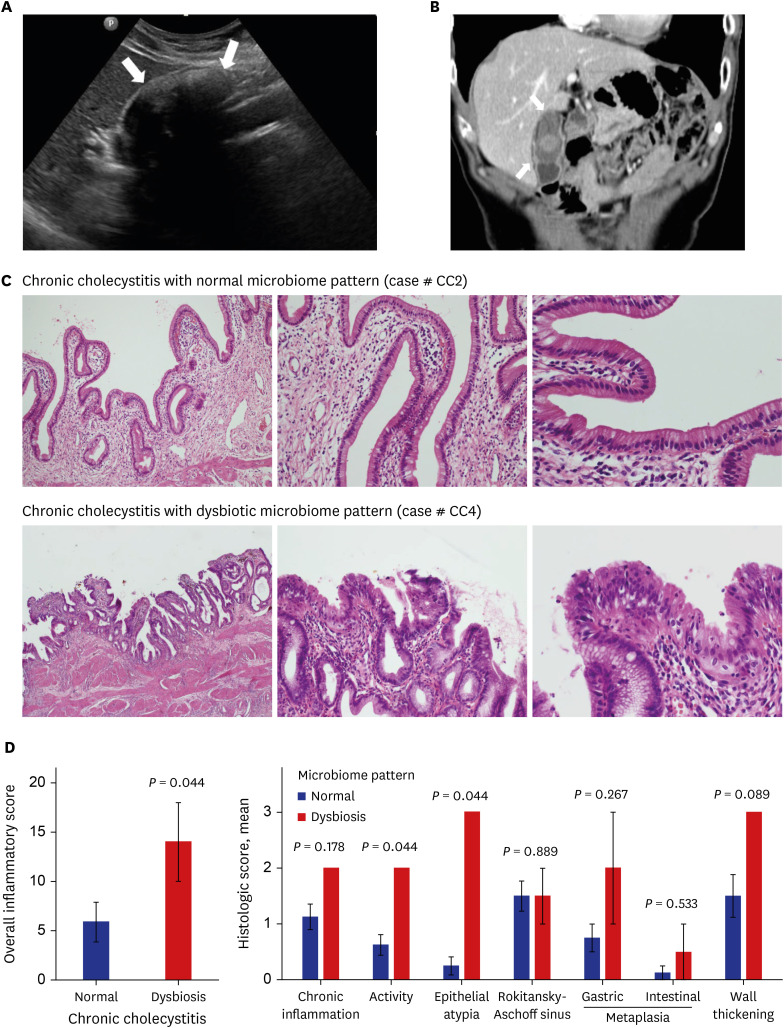
Fig. 5
Radiologic and pathologic findings of CC patients with or without a dysbiotic microbiome. B-mode ultrasound (A) and coronal computed tomography scan (B) obtained on portal phase show the stone with the largest dimension (4.4 cm) in the gallbladder of a 77-year-old man. (C) CC with a gallbladder cancer-associated microbiome pattern reveals severe infiltration of lymphoplasmacytic cells and neutrophils and shallow mucosal erosion. On higher magnification, the epithelium shows enlarged, hyperchromatic, crowded nuclei with prominent nucleoli. However, CC with a normal microbiome pattern shows nonspecific chronic inflammation without epithelial atypia. (D) The overall histologic score of CC with dysbiosis is significantly higher than the normal, especially regarding the degree of activity and epithelial atypia.
CC = chronic cholecystitis.
Pathological findings of CC with or without a dysbiotic microbiome
The detailed histopathological features of each chronic cholecystectomy specimen are described in Supplementary Table 5. Diffuse epithelial atypia (which is characterized by enlarged hyperchromatic nuclei with distinct nucleoli, slight loss of cell polarity, and crowding) was observed in patients with a dysbiotic microbiome. Active lesions (characterized by vascular congestion, neutrophilic infiltration, and erosion) were also more frequent in patients with a dysbiotic microbiome. There was no difference between the groups with regard to microbiome pattern and other histological features, such as the degree of chronic inflammation, metaplasia, Rokitansky-Aschoff sinuses, and wall thickening. However, the overall inflammatory score of patients with CC and a dysbiotic microbiome was significantly higher than the score of those with a normal microbiome (Mann-Whitney U test, P = 0.044) (Fig. 5C and D).
DISCUSSION
The major finding from our study is the different microbiota of bile in healthy control, patients with CC, and patients with gallbladder cancer. No meaningful bacteria were identified in the bile samples from the healthy control group, while four out of ten bile samples (40%) from CC patients and all samples (100%) from GBC patients showed dysbiosis (Fig. 2).
Bile in a normal biliary system has been considered sterile for a long time. Recently, several studies have suggested the existence of a normal bile microbiota including Firmicutes, Bacteroidetes, Actinobacteria, and Proteobacteria.1329 The current study tried to identify the microbiota of the bile collected from the normal GBs, but no meaningful bacteria were detected, as assumed previously.
The predominant dysbiotic bacteria in CC were those belonging to the Enterobacteriaceae family, which includes Escherichia coli, Klebsiella, Salmonella, Shigella, and Citrobacter.30 These comprise a major pathogenic group in the human symbiotic microbial ecosystem and are commonly observed in various physical illnesses including gastrointestinal, and urinary tract infections.31 Among them, Citrobacter was one of the predominant bacteria found in the CC group in our study. Citrobacter is isolated from various clinical samples, such as urine, sputum, and other body fluids, and has been reported to induce infections in various organs.32 Especially in our study, C. braakii and C. freundii were the two dominant species identified in CC patients. C. freundii was previously reported to be associated with GB disease, especially in acute cholecystitis.33 Other bacteria identified in CC were Klebsiella, Enterococcus, Enterobacter, Escherichia, Kosakonia, Yokenella, and Bacteroides. In the literature, there are several studies on bacteria in CC, but most of the results are combined with other GB diseases, mainly acute cholecystitis, and could not be isolated. Salmonella and Helicobacter species were reported to be associated with CC, and the subsequent development of GBC.343536 However, these bacteria were not found in our samples, probably due to the previous use of antibiotics.
The predominant dysbiotic bacteria in the bile from GBC patients in our study were Klebsiella, Escherichia, Enterococcus, Enterobacter, Haemophilus, and Streptococcus, which belong to the Enterobacteriaceae or Streptococcaceae families. The results were relatively consistent with the GBC samples, where Enterobacter, Escherichia, and Klebsiella were significantly abundant as compared to those found in the normal bile samples. Although there were variations among the reports, most of these bacteria have been frequently reported in GBC.3738 Few cases with Streptococcus infection and coexisting GBC have also been reported.3940 However, Haemophilus has not been identified in GBC before, but still, several studies have reported the isolation of the bacteria in biliary tract infection.4142 Interestingly, Salmonella or Helicobacter, which were considered as one of the causative bacteria, were not identified in the bile from GBC patients. However, several previous studies with bile samples from GBC could not find them either.3743 The differences in the results might be due to the diversity in the samples/methods used, populations, or small numbers of participants in these studies. The detection rates of bile bacteria in GBC were 40–81% in previous studies, but the detection rate in our study was 100%, therefore sensitivity may not be a concern.3744
One of the most important findings in our study was that the prevalence of Klebsiella significantly increased in the order of normal, CC, and GBC. The bacteria with the highest coverage among Klebsiella was K. oxytoca (84.24%). Several studies have reported K. oxytoca bacteremia association with biliary infection,454647 and few studies have reported the K. oxytoca in bile samples from GBC.37 It was also demonstrated that K. oxytoca act as a gut pathobiont that increase the gut permeability and influence in cancer progression on a murine model.48 Our result suggests the possible relation of Klebsiella in the process of GB carcinogenesis. Further studies on the pathogenic properties of Klebsiella cytotoxin are required to support this result.
We also compared the clinicopathological differences between the normal and dysbiotic microbial groups among CC patients. CC with a dysbiotic microbiota correlated with specific findings, such as large stones (more than 3 cm in diameter) in CT scan and marked epithelial atypia in pathology. Gallstone, which is found in approximately 85% of GBC patients, is suggested to be one of the most important risk factors of GBC, and the risk of GBC is reported to correlate with the increasing size of gallstones.495051 Its mechanism is not completely understood, but it is presumed that chronic inflammation in the GB caused by gallstones leads to epithelial damage and then to GBC by carcinogenesis.52 Patients with CC and epithelial atypia, therefore, have a greater potential for GBC development, as epithelial atypia can lead to malignant transformation.5354
Detection of certain bacteria does not suggest whether these bacteria are the cause or effect of inflammation or cancer. However, accumulating evidence points out that chronic inflammation and microbiota dysbiosis contribute to the carcinogenesis. There is some evidence in our study that suggests the relation between the bacteria we identified and GB carcinogenesis. First, the bacteria we identified in bile from GBC were rather consistent among the patients. Especially, the CLR of Klebsiella was significantly elevated in CC and gallbladder cancer in order that its concentration may have a role in carcinogenesis. Second, the bacteria we identified were also found in previous literature to cause CC and GBC. Third, the groups with microbiota dysbiosis showed pathologic and radiologic findings that are risk factors or premalignant lesions of GBC, respectively. Although we were unable to prove that the bacteria we identified are the cause of the pathologic phenotype, we could suggest from this evidence that these bacteria could be a possible candidate for GB carcinogenesis, and it is our next step to find the connection between the pathogen and the disease.
There are several limitations to our study. Because of a small number of participants and low signal detection rate in normal control, the representativeness of each group is to be considered. There were several differences in the baseline characteristics between healthy controls and patients with GB diseases, and a substantial person-to-person variation could not be avoided. The isolation of bacteria does not imply that they are pathogenic, therefore, follow-up studies are required to elucidate the possible link between the revealed bacteria and pathology.
Using metagenomic sequencing, we identified the bile microbiome of GB diseases and the correlations between their dysbiosis and clinical features. The predominant dysbiotic bacteria in the bile from CC and GBC mainly belonged to the Enterobacteriaceae family. We found a significant stepwise increase in Klebsiella in the bile of GBC patients as compared to the bile of normal and CC patients. Additionally, CC patients with dysbiotic bile tended to have larger gallstones and epithelial atypia, which are risk factors for GBC and potential for the development of GBC, respectively. Together, our results suggest a link between microbial disturbance and GB carcinogenesis. Further studies are needed to support our results.
 XML Download
XML Download