

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Cholesterol is necessary for good health. However, hypercholesterolemia, defined as an abnormally increased level of cholesterol in blood, is a serious health condition. It is a major risk factor for the development of cardiovascular diseases, such as atherosclerosis and its complications. In addition, many studies suggest an association of dietary cholesterol intake with the risk of nonalcoholic fatty liver disease (NAFLD), a broad spectrum of liver diseases, ranging from simple steatosis to steatohepatitis, fibrosis, cirrhosis, and liver cancer [12345]. Although most patients with NAFLD are obese, NAFLD is also observed in non-obese subjects [1]. Epidemiologic studies suggest that a high-cholesterol diet (HCD) is a critical factor in non-obese NAFLD [12]. In a small Italy-based study, normal-weight patients with steatohepatitis showed higher cholesterol consumption than that of BMI-matched healthy controls [3]. Yasutake et al. [1] also reported that cholesterol intake in non-obese NAFLD patients was notably high compared to that in obese NAFLD patients and healthy volunteers, although dietary intake levels of total energy, fat, and carbohydrate were not excessive in the non-obese patients. Animal studies using cholesterol-rich diets support the results obtained in non-obese NAFLD patients [45]. C57BL/6J mice fed an HCD (containing 1.25% cholesterol and 0.5% cholic acid) developed progressive steatosis, inflammation, and fibrosis without obesity [4]. Recently, Tu et al. [5] also demonstrated that HCD-induced non-obese NAFLD might be distinct from obese NAFLD occurring as a consequence of metabolic syndrome. Moreover, patients with NAFLD are at an increased risk of cardiovascular disease, and NAFLD is proposed as an independent risk factor for cardiovascular disease [67].
Nobiletin (5,6,7,8,3′,4′-hexamethoxyflavone, NOB) is a flavonoid present in the peel of citrus fruits such as Citrus depressa (shiikuwasa), Citrus sinensis (oranges), and Citrus limon (lemons) [8]. It has been reported that NOB has various pharmacological activities, such as anti-inflammation, antioxidation, anti-cancer, and neuroprotection effects [8]. Hepatoprotective, cardioprotective, and metabolically beneficial properties of NOB have also been demonstrated [91011]. In vitro, NOB inhibited hepatic lipogenesis in HepG2 hepatocytes via modulation of the AMPK signaling pathway and exerted cardiovascular protective effects by preventing the oxidized low-density lipoprotein (oxLDL)-mediated expression of Tissue Factor in human endothelial cells through the inhibition of nuclear factor-κB [910]. In vivo studies support a link between NOB and anti-metabolic effects [111213]. NOB (0.1% or 0.3%) prevented dyslipidemia, hepatic triglyceride accumulation, and atherosclerotic lesion development in high-fat diet (HFD, 42 kcal% fat, no added cholesterol or cholic acid)-fed low-density lipoprotein receptor (Ldlr)−/− mice [11]. In other research conducted in their laboratory, Ldlr−/− mice were initially fed an HFD (42 kcal% fat, 0.2% cholesterol) over 12 weeks and received an HFD supplemented with 0.3% NOB for the subsequent 12 weeks [12]. NOB attenuated obesity, insulin resistance, hyperlipidemia, and hepatic steatosis, and it favorably altered aortic sinus atherosclerotic plaque composition [12]. Similarly, our previous study demonstrated that NOB supplementation (0.02%, approximately 17 mg/kg body weight/day) for 16 weeks attenuated dyslipidemia, hepatic steatosis, insulin resistance, and inflammation without altering adiposity in HFD (45 kcal% fat, no added cholesterol or cholic acid)-induced obese mice [13]. However, there is limited research investigating the effect of long-term supplementation with low-dose NOB on HCD-induced hypercholesterolemia and non-obese NAFLD.
In the present study, we hypothesized that long-term supplementation with low-dose NOB might exert protective effects against HCD-induced hypercholesterolemia and non-obese NAFLD by regulating lipogenesis and fatty acid oxidation in the liver, and these beneficial effects might be associated with the anti-inflammatory and cardiovascular protective effects of NOB. Therefore, we investigated the effects of NOB on plasma levels of lipids and inflammatory and atherosclerosis markers, as well as on hepatic morphology and lipid content in HCD-fed C57BL/6J mice. The enzyme activities and messenger RNA (mRNA) expression levels of genes involved in lipid metabolism were also evaluated. In particular, the present study focused on evaluating the effects of NOB on hepatic cholesterol metabolism, including cholesterol synthesis, esterification, and influx.
Go to : 
MATERIALS AND METHODS
Animals and experimental diets
Four-week-old male C57BL/6J mice were obtained from Jackson Laboratories (Bar Harbor, ME, USA) and housed under standard conditions with free access to chow and water. All animals were acclimated for 1 week before use. At 5 weeks of age, they were randomly divided into three groups (n = 12 in each group). The first group was fed a normal diet (ND). The second group was considered the negative control group and was fed only an HCD (D12336; Research Diets, New Brunswick, NJ, USA). The third group was fed the HCD and received dietary supplementation with NOB (0.02%). NOB was isolated from the peels of shiikuwasa (C. depressa) by performing methanol extraction followed by two chromatographic steps; its purity was verified by nuclear magnetic resonance and mass spectroscopy, as previously described [13]. The HCD contained 16% fat (5% soybean oil, 7.5% cocoa butter, 3.5% coconut oil; 35 kcal% fat), 1.25% cholesterol, and 0.5% cholic acid. Mice were provided access to food and water ad libitum during the 20-week study period. All animal procedures related to the animal studies were approved by the Ethics Committee at Kyungpook National University (approval No. KNU-2014-45).
Food consumption and body weight were measured daily and weekly, respectively. At the end of the experimental period, the mice were anesthetized with isoflurane (5 mg/kg body weight; Baxter, USA) following a 12-h fast. Blood samples were collected from the inferior vena cava into a heparin-coated tube for plasma biochemical analysis. Liver and white adipose tissue were excised, weighed, and snap-frozen in liquid nitrogen. All tissues were stored at −70°C until further analyses.
Plasma biochemical analysis
The plasma levels of total cholesterol (TC) and triglycerides were determined using enzymatic kits (Asan, Seoul, Republic of Korea). The levels of high-density lipoprotein (HDL) and low-density lipoprotein (LDL)/very-LDL (VLDL) in plasma were measured using an HDL and LDL/VLDL-cholesterol assay kit (Abcam, Cambridge, MA, UK). Plasma C-reactive protein (CRP; R&D systems, Minneapolis, NE, USA) and oxLDL (MyBioSource, San Diego, CA, USA) levels were assessed using commercial assay kits. Plasma levels of adipocytokines (adiponectin, plasminogen activator inhibitor-1 [PAI-1], interleukin [IL]-1β, and IL-6) were determined using a multiplex detection kit (Bio-Rad, Hercules, CA, USA).
Hepatic lipid analyses
Hepatic lipids were extracted as previously described [14], and dried lipid residues were dissolved in 1 mL of ethanol for triglyceride and cholesterol assays. Triton X-100 and a sodium cholate solution in distilled water were added to 200 μL of the dissolved lipid solution for emulsification. The hepatic triglyceride and cholesterol contents were analyzed with the same enzymatic kit used for the plasma analysis.
Enzyme analyses
Cytosolic, mitochondrial, and microsomal fractions were prepared from liver homogenate using differential centrifugation to determine the activities of lipid-regulating enzymes. The 3-hydroxy-3-methylglutaryl (HMG)-coenzyme A (CoA) reductase and acyl-CoA:cholesterol acyltransferase (ACAT) activities in the microsomal fraction were determined by a procedure adapted from those of Shapiro et al. [15] and Erickson et al. [16], respectively. Cytosolic fatty acid synthase (FAS) activity was measured by monitoring the malonyl CoA-dependent oxidation of NADPH based on the absorbance of the samples at 340 nm [17]. Mitochondrial carnitine palmitoyltransferase (CPT) activity was determined using a spectrophotometric assay that measured the CoA-SH release from palmitoyl-CoA [18]. Mitochondrial fatty acid β-oxidation was measured by monitoring the reduction of NAD+ to NADH in the presence of palmitoyl-CoA by applying a previously described method [19]. Protein concentration in each fraction was estimated by using the Bradford method [20]. Plasma paraoxonase activity was spectrophotometrically assayed using the method described by Mackness et al. [21].
Isolation of total RNA and quantitative real-time reverse transcription polymerase chain reaction (qRT-PCR) analysis
Total RNA was isolated from liver using TRIZOL reagent (Invitrogen Life Technologies, Grand Island, NY, USA). RNA integrity for each sample was evaluated by using the Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA). The complementary DNA was synthesized using 1 µg RNA and a QuantiTect® reverse transcription kit (Qiagen, Hilden, Germany). qRT-PCR was carried out on a CFX96TM real-time system (Bio-Rad) using the SYBR Green qRT-PCR kit (Qiagen). The mRNA levels of each target gene were normalized to that of GAPDH mRNA. The relative gene expression levels were calculated according to the 2−ΔΔCT method.
Histological analysis
Liver tissues were fixed in 10% formalin solution, dehydrated, embedded in paraffin, and cut into 4-μm-thick sections. Cross-sections of these tissues were stained with hematoxylin and eosin. Stained areas were viewed using an optical microscope (Nikon, Tokyo, Japan) and a magnifying power of 200×.
Statistical analysis
The results are presented as mean ± SE values. Differences between 2 groups (ND vs. HCD and HCD vs. HCD + NOB) were determined using Student's t-test. Initially, to determine whether there is statistical evidence that the diet-associated means are significantly different, Student's t-test was applied to the means of the ND and HCD groups. Also, in order to determine the effect of NOB on HCD-induced hypercholesterolemia and NAFLD, the Student's t-test was used to compare the mean values of HCD-fed mice receiving or not receiving NOB supplementation. Test result P-values of less than 0.05 were considered statistically significant. Statistical analysis was performed using SPSS statistical software (version 11.0; SPSS Inc., Chicago, IL, USA).
Go to :
RESULTS
NOB suppresses HCD-induced weight loss
HCD-fed mice showed significantly lower food intake, body weight, fat mass (total weight of all white adipose tissue depots including epididymal, perirenal, retroperitoneal, mesenteric, subcutaneous, and interscapular white adipose tissue), and food efficiency ratio (FER) compared to those of ND-fed mice. However, there was no significant difference in energy intake between the ND and HCD groups (Fig. 1). In HCD-fed mice, dietary NOB supplementation did not affect the amount of food consumed, energy intake, and fat mass (Fig. 1A, B, and E). However, NOB tended to increase final body weight (P = 0.51), and NOB-supplemented mice showed significantly increased body weight gain and FER compared to HCD control mice (Fig. 1C, D, and F).
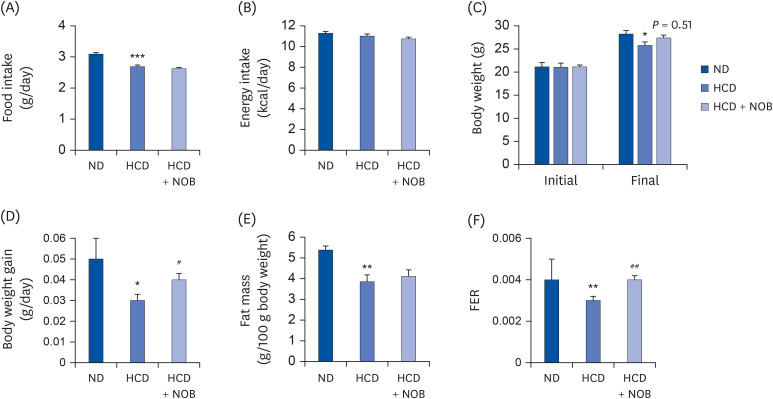 | Fig. 1Effect of NOB on food intake (A), energy intake (B), body weight (C, D), fat mass (E), and FER (F) in HCD-fed mice. Values are presented as mean ± SE (n = 12). Values are significantly different between the groups, according to Student's t-test.ND, normal diet; HCD, high-cholesterol diet (35 kcal% fat, 1.25% cholesterol, 0.5% cholic acid); HCD + NOB, high-cholesterol diet plus nobiletin (0.02%); FER, food efficiency ratio.
*P < 0.05, **P < 0.01, ***P < 0.001, ND vs. HCD; #P < 0.05, ##P < 0.01, HCD vs. HCD + NOB.
|
NOB decreases the levels of circulating cholesterol, CRP, oxLDL, inflammatory markers, and PAI-1 but increases the circulating adiponectin level and paraoxonase activity
Over 20 weeks, plasma TC levels were significantly increased in HCD-fed mice compared to those of the ND-fed mice (Fig. 2A). In addition, mice fed with HCD showed a significantly higher ratio of plasma LDL/VLDL-cholesterol to TC, and the HDL-cholesterol/TC ratio and triglyceride levels were lower in the HCD group than those in the ND group (Fig. 2B and C). In HCD-fed mice, NOB supplementation significantly lowered the plasma TC levels compared to the HCD control group level (Fig. 2A). Moreover, the mean ratio of plasma LDL/VLDL-cholesterol to TC in NOB-supplemented mice was significantly lower than that in HCD control mice, whereas NOB supplementation markedly increased the HDL-cholesterol/TC ratio but did not significantly affect the plasma triglyceride level (Fig. 2B and C).
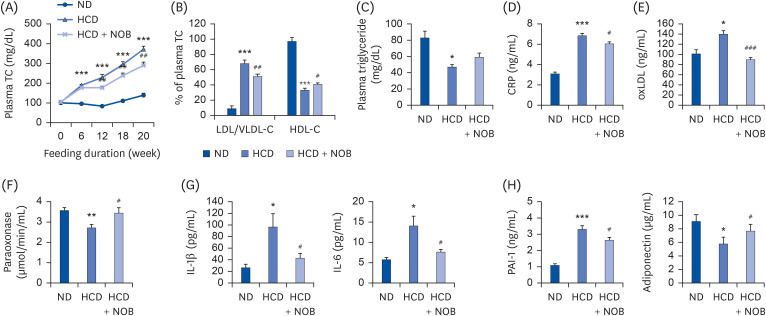 | Fig. 2Effect of nobiletin on plasma levels of TC (A), ratios of LDL/VLDL- and HDL-cholesterol to TC (B), and plasma levels of triglyceride (C), CRP (D), oxLDL (E), paraoxonase (F), and adipocytokines (G, H) in HCD-fed mice. Values are presented as mean ± SE (n = 12). Values are significantly different between the groups, according to Student's t-test.TC, total cholesterol; ND, normal diet; HCD, high-cholesterol diet (35 kcal% fat, 1.25% cholesterol, 0.5% cholic acid); HCD + NOB, high-cholesterol diet plus nobiletin (0.02%); LDL/VLDL-C, low-density lipoprotein/very low-density lipoprotein-cholesterol; HDL-C, high-density lipoprotein-cholesterol; CRP, C-reactive protein; oxLDL, oxidized low-density lipoprotein; IL, interleukin; PAI-1, plasminogen activator inhibitor-1.
*P < 0.05, **P < 0.01, ***P < 0.001, ND vs. HCD; #P < 0.05, ##P < 0.01, ###P < 0.001, HCD vs. HCD + NOB.
|
HCD feeding also caused significant increases in the levels of plasma markers of inflammation and atherosclerosis, such as CRP, oxLDL, IL-1β, IL-6, and PAI-1, compared to those from ND feeding. Conversely, the activity of plasma paraoxonase, an HDL-associated enzyme exhibiting potentially anti-atherogenic properties, was markedly lowered in HCD-fed mice compared to that in ND-fed mice (Fig. 2D-H). Dietary NOB supplementation normalized the HCD-induced changes to these markers (Fig. 2D-H). Furthermore, plasma adiponectin levels were significantly lower in HCD-fed mice than those in ND-fed mice, and NOB supplementation markedly increased the plasma adiponectin levels compared to those in the HCD group (Fig. 2H).
NOB decreases liver weight and hepatic lipid accumulation
Hepatic cholesterol and triglyceride contents, as well as liver weight, were higher in HCD-fed mice than in ND-fed mice (Fig. 3A and B). In contrast, NOB-supplemented mice showed significantly reduced liver weight and hepatic cholesterol and triglyceride contents compared to those of the HCD control group. Morphological analyses of liver tissues also indicated that lipid droplet accumulation was more pronounced in HCD-fed mice than in ND-fed mice; however, NOB supplementation markedly decreased hepatic lipid accumulation compared to that in the HCD-fed mice (Fig. 3C). Overall, dietary NOB supplementation might ameliorate HCD-mediated hepatic steatosis in mice.
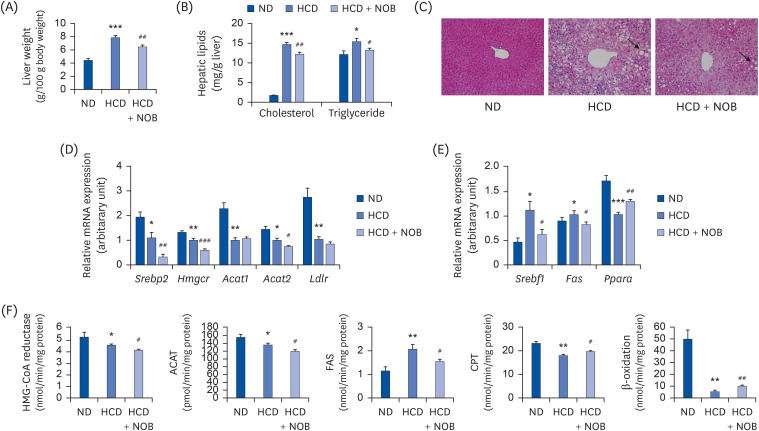 | Fig. 3Effect of NOB on liver weight (A), hepatic lipid content (B), liver morphology (C), expression of hepatic lipid metabolism-related genes (D, E), and activities of hepatic lipid metabolism-related enzymes (F) in HCD-fed mice. (A, B, D-F) Values are presented as mean ± SE (n = 12). Values are significantly different between the groups, according to the Student's t-test. (C) Representative photomicrographs of livers are shown at 200× magnification.ND, normal diet; HCD, high-cholesterol diet (35 kcal% fat, 1.25% cholesterol, 0.5% cholic acid); HCD + NOB, high-cholesterol diet plus nobiletin (0.02%); mRNA, messenger RNA; HMG-CoA reductase, 3-hydroxy-3-methylglutaryl-CoA reductase; ACAT, acyl-CoA:cholesterol acyltransferase; FAS, fatty acid synthase; CPT, carnitine palmitoyltransferase.
*P < 0.05, **P < 0.01, ***P < 0.001, ND vs. HCD; #P < 0.05, ##P < 0.01, ###P < 0.001, HCD vs. HCD + NOB.
|
NOB regulates expressions of lipid metabolism-related genes and enzyme activity in liver
To determine how NOB ameliorated HCD-induced hepatic steatosis, we examined the mRNA expression levels of genes and the activities of enzymes involved in hepatic cholesterol and triglyceride accumulation. HCD feeding led to a significant decrease in the mRNA expression of hepatic genes involved in cholesterol synthesis, esterification, and influx (Srebp2, Hmgcr, Acat1, Acat2, Ldlr) compared to those in the ND group (Fig. 3D). In addition, the mRNA expression levels of hepatic Srebf1, a key lipogenic transcription factor, and its target gene Fas were higher in the HCD group, whereas those of hepatic Ppara, a major transcription factor involved in fatty acid β‐oxidation, were lower in the HCD group compared with those in the ND group (Fig. 3E). Mice that received NOB supplementation showed significantly decreased mRNA expression of genes involved in cholesterol synthesis and esterification (Srebp2, Hmgcr, Acat2) as well as in lipogenesis (Srebf1 and Fas) in the liver, along with the upregulation of hepatic Ppara mRNA expression, compared to the expression levels in the HCD negative control group (Fig. 3D and E). Similar to the trends observed in gene expression, the activities of HMG-CoA reductase, ACAT, CPT, and β-oxidation were significantly lower in the HCD group than in the ND group (Fig. 3F). In contrast, hepatic FAS activity was significantly higher in the HCD group than in the ND group. In HCD-fed mice, NOB supplementation significantly decreased the activities of hepatic HMG-CoA reductase, ACAT, and FAS (Fig. 3F). Moreover, NOB significantly increased CPT and β-oxidation activities compared to those in the HCD group (Fig. 3F).
Go to :
DISCUSSION
The HCD-fed C57BL/6J mouse utilized in the present study is a commonly used animal model of hypercholesterolemia and atherosclerosis [22]. Unlike most mouse strains resistant to developing hypercholesterolemia and atherosclerosis, even on an HCD, the HCD-fed C57BL/6 mouse exhibits an approximate 50% reduction in the plasma HDL-cholesterol level [22]. Similarly, in the present study, HCD-fed C57BL/6J mice demonstrated significantly higher TC and LDL/VLDL-cholesterol to TC ratio values and a lower HDL-cholesterol/TC ratio. Moreover, HCD-fed mice developed NAFLD and showed a significantly lower weight gain compared to that of ND-fed mice, although the daily energy intake was similar in both groups. These results are consistent with those of a previous study [4], which demonstrated that HCD-fed mice are not obese but show hepatomegaly and NAFLD. Since an atherogenic diet high in cholesterol and cholic acid can induce toxicity symptoms like weight loss [23], it seems that the loss of body weight and fat mass observed in HCD group may be the result of a toxic effect and dietary growth inhibition.
The present study showed that dietary supplementation with low-dose NOB (0.02%) for 20 weeks suppressed HCD-induced weight loss and decreased TC plasma levels in mice fed an HCD. In addition, NOB markedly decreased the ratio of plasma LDL/VLDL-cholesterol to TC but increased the HDL-cholesterol/TC ratio compared to that of the HCD control group. Similar to our results, NOB was shown to decrease the circulating levels of VLDL and LDL in vitro [24], and HFD-fed aged mice supplemented with NOB (0.1%) showed reductions in serum LDL/VLDL-cholesterol levels and the LDL/HDL ratio [25]. Recently, Morrow et al. [26] also demonstrated that plasma TC and LDL-cholesterol levels were decreased by NOB (0.3%) in C57BL/6J mice fed an HFD (42 kcal% fat, 0.2% cholesterol).
The beneficial effects of NOB on HCD-induced hypercholesterolemia may contribute to protection against cardiovascular disease. Hypercholesterolemia is generally associated with increased levels of oxLDL [27], which leads to the activation of pro-inflammatory and atherogenic cytokines, thereby contributing to the progression of atherogenesis [28]. Plasma oxLDL is considered a strong predictor of atherosclerotic cardiovascular disease [29]. In addition to affecting oxLDL, NOB diminishes the HCD-mediated upregulation of circulating inflammatory markers, like CRP, IL-6, and IL-1β, which have been shown to independently predict cardiovascular disease [3031323334]. In particular, circulating CRP is stable over long periods, has no diurnal variation, and has been suggested to be a stronger predictor of cardiovascular events than the LDL-cholesterol level [3031]. Taken together, NOB might exert cardiovascular protective effects by preventing oxLDL formation and decreasing the plasma levels of the pro-inflammatory and atherosclerosis markers CRP, IL-6, and IL-1β. These findings are supported by a decreased level of PAI-1, increased level of adiponectin, and increased activity of paraoxonase, an HDL-associated enzyme that protects against LDL oxidation, observed in the plasma of NOB-supplemented mice. A high level of PAI-1, a major regulator of the fibrinolytic system, is associated with an increased cardiovascular risk of arterial and thrombotic disease [35], whereas circulating adiponectin exerts protection against cardiovascular disease [36]. In animal studies using either pharmacological or genetic approaches, inhibition of PAI-1 is suggested to be a therapeutic option for cardiovascular protection, and the elevation of plasma adiponectin alleviates atherosclerosis [3637]. In addition, several studies have demonstrated a protective role of paraoxonase in cardiovascular diseases, such as atherosclerosis and ischemic stroke [3839].
The liver is the principal organ for cholesterol homeostasis. Cholesterol is synthesized primarily in the liver and transported to other tissues via the blood in the form of lipoproteins. Hepatic cholesterol synthesis, storage by esterification, uptake, and excretion have important roles in whole-body cholesterol homeostasis, and losing control of any of these processes results in hypercholesterolemia and increases the risk for cardiovascular disease [40]. HMG-CoA reductase is responsible for cholesterol synthesis in the liver [40], and ACAT converts cholesterol into its storage form, cholesteryl esters [40]. In mammals, there are two isoforms of ACAT; Acat1 is a ubiquitous gene, whereas Acat2 is primarily located in liver and intestine. The hepatic Acat2 synthesizes cholesteryl esters for incorporation into VLDL and provides cholesteryl ester for the formation of cytoplasmic lipid droplets, a storage method when liver cholesterol is abundant [41]. Deletion of liver-specific Acat2 results in resistance to hypercholesterolemia and hepatic lipid accumulation induced by a diet high in fat and cholesterol in mice [42], while Acat1-deficient mice showed no apparent effects on plasma cholesterol level and cholesterol esterification activity [43], indicating a specialized role of Acat2 in cholesteryl ester synthesis in the liver. Interestingly, in the present study, NOB significantly downregulated the hepatic mRNA expression of Hmgcr and Acat2, although it did not alter hepatic Acat1 mRNA expression. Moreover, the mRNA expression of hepatic Srebp2, a primary transcriptional factor for the activation of Hmgcr [44], was downregulated, and NOB inhibited the corresponding enzyme activities. Therefore, it seems possible that the inhibition of cholesterol synthesis and esterification might reduce the availability of hepatic cholesterol for VLDL formation and contribute to decreased secretion of VLDL, which consequently ameliorates HCD-induced hypercholesterolemia.
Furthermore, reduced cholesterol synthesis and esterification through the downregulation of hepatic Srebp2, Hmgcr, and Acat2 could contribute to the attenuation of HCD-induced NAFLD observed in NOB-supplemented mice, since disturbed hepatic cholesterol homeostasis is relevant to the pathogenesis of NAFLD [45]. Expression of hepatic Hmgcr and Srebp2 is increased in NAFLD patients [3], and liver-specific inhibition of Acat2 with antisense oligonucleotides decreases the accumulation of neutral lipids (cholesteryl ester and triglyceride) in HCD-fed mice [46]. Another potential mechanism underlying the protective effects of NOB against NAFLD might be associated with decreased de novo lipogenesis and increased fatty acid oxidation in the liver. In a previous study, feeding C57BL/6J mice with an HCD (1.25% cholesterol, 0.5% cholic acid) not only upregulated the hepatic mRNA expression of genes involved in de novo lipogenesis (Srebf1 and Fas) but also downregulated the hepatic mRNA expression of genes associated with the mitochondrial fatty acid oxidation pathway (Ppara and Cpt1a), contributing to the pathogenesis of non-obese NAFLD [6]. Similarly, the present study demonstrated markedly increased expression and activity levels of de novo fatty acid synthesis-related genes and enzymes, respectively, and decreased the expression and activities of fatty acid oxidation-related genes and enzymes, respectively, in the liver in response to the HCD. Notably, NOB supplementation normalized these HCD-induced changes in the expression and activities of these fatty acid synthesis- and β-oxidation-related genes and enzymes, respectively, in the liver. These results indicate that the protective effects of NOB against HCD-induced NAFLD might be partly due to decreased lipogenesis and increased fatty acid oxidation in the liver, along with the regulation of cholesterol metabolism.
In a previous study, we demonstrated that NOB can protect against dyslipidemia and NAFLD in HFD (no added cholesterol or cholic acid)-induced obese mice [13]. However, the mechanisms underlying its protective effects against HCD (containing high-cholesterol and cholic acid)-related metabolic dysfunction, such as hypercholesterolemia and non-obese NAFLD, remain unclear. Therefore, the present study focused on evaluating the effects of NOB on hepatic cholesterol metabolism (including cholesterol synthesis, esterification, and influx) and hypercholesterolemia-associated plasma biomarkers (including CRP, oxLDL, IL-1β, IL-6, PAI-1, adiponectin and paraoxonase) in HCD-fed C57BL/6J mice, a commonly used animal model of hypercholesterolemia and atherosclerosis [22]. Our novel findings reveal that NOB could protect against hypercholesterolemia and non-obese NAFLD by inhibiting mRNA expression of hepatic genes and activities of cholesterol synthesis and esterification, along with inhibition of fatty acid synthesis and promotion of fatty acid oxidation. Also, these effects were associated with the amelioration of inflammation and the regulation of plasma levels of atherosclerosis-associated cardiovascular markers.
In conclusion, the present study demonstrates, for the first time, that long-term supplementation of low-dose NOB might attenuate HCD-induced hypercholesterolemia and NAFLD by regulating cholesterol synthesis and esterification, de novo lipogenesis, and fatty acid oxidation in the liver. In addition, NOB supplementation can decrease pro-inflammatory and atherosclerosis marker levels and increase adiponectin level and paraoxonase activity in plasma, suggesting that it might exert cardioprotective effects. Taken together, the results support the suggestion that NOB can potentially ameliorate hypercholesterolemia, cardiovascular disease, and NAFLD.
Go to :
 XML Download
XML Download