

 PDF
PDF Citation
Citation Print
Print



Introduction
Porotic hyperostosis (PH) refers to the expansion of the diploë of the outer table cranial vault, producing sieve-like porosities. When shown on X-ray, PH is marked by the ‘hair-on-end’ appearance [1] with thin vertical striations or the accentuated vertical trabeculae between the inner and outer tables of the skull. PH may also be observed in the orbital roof, a condition known as cribra orbitalia (CO). The term CO is becoming progressively popular and treated as synonym in the modern literature. Both PH and CO are commonly seen in archaeological collections, and are regarded as a pathological condition and used as indicators of nutritional status in prehistoric and historic contexts.
Considering its etiology, both PH and CO are regarded as a manifestation of anemia [2]. Anemia may have an acquired origin resulting from malnutrition such as iron deficiency, vitamin B deficiency, magnesium deficiency, or from a genetic origin such as thalassemia [3]. Thalassemia is an inherited blood disorder characterized by decreased production of hemoglobin. It is caused by a mutation of one of the genes that encode the hemoglobin chains, leading to faulty hemoglobin production, ineffective erythropoiesis, which eventually results in anemia. PH occurs in response to anemia regardless of its origin due to marrow hyperplasia (increased hematopoietic cell number) and hypertrophy (increased cell size), resulting in high level of erythropoiesis to keep up with chronic destruction of red blood cells. These events take in conjunction with resorption of the outer table producing coral-like porosities.
Epidemiologically, PH due anemia is considered very rare in the modern context and is more closely related to genetically acquired anemia, especially when global malnutrition has immensely declined. Yet, not all cases of anemia would produce massive marrow hypertrophy that is responsible for PH, and only those cases with marrow hypertrophy prolonged erythropoiesis can be linked to PH [3]. A recent review by Cole and Waldron [4] revealed that PH is an extremely rare condition with a pooled prevalence of only 0.08% in the relatively modern populations. In the present study, we report the gross morphology of the two skulls affected by extensive PH, one of which belonged to a clinically diagnosed beta thalassemia patient, and the other belonged to a patient with unspecified type of thalassemia. The radiologic appearance of the two skulls was examined by X-ray and the significance of our findings are discussed.
Go to : 
Case Report
This study was approved by the Institutional Review Board of the Faculty of Medicine, Khon Kaen University (approval number: HE631508). During the routine preparation of skeletal remains at the Unit of Human Bone Warehouse for Research, Department of Anatomy, Faculty of Medicine, Khon Kaen University, we encountered two skulls with aberrant gross morphology. The body of the donors of the two skulls were donated to our department with an informed consent. Skulls were photographed and examined by X-ray (Toshiba KXO-15E, Toshiba Electron Tubes & Devices).
The first skull (Fig. 1A-C) belonged to a woman aged 41-year-old (height 157 cm. and weight 38 kg.) who had previously been diagnosed with beta thalassemia (skeleton number 51/0149). Excessive PH was seen throughout the skull and the maxilla. Coral-like vertical striations were prominent and symmetrical. Severe CO was observed on the orbital roof and the superciliary arch (brow ridge) on both sides of the orbit without convexity of the orbital roof (Fig. 1A) Temporal bone of both sides was unaffected by the porotic lesions. The second skull (Fig. 1D-F) belonged to a 35-year-old man (height 160 cm. and weight 42 kg.), who had previously been diagnosed with unspecified thalassemia (skeleton number 59/3161). This skull exhibited porosities in the frontal lobe and the parietal lobe. CO was not observed and the orbital roof of both orbits was totally intact (Fig. 1D). Sagittal suture was not visible. Coronal suture was only apparent on the temporal sides and the temporal bone of both sides were spared.
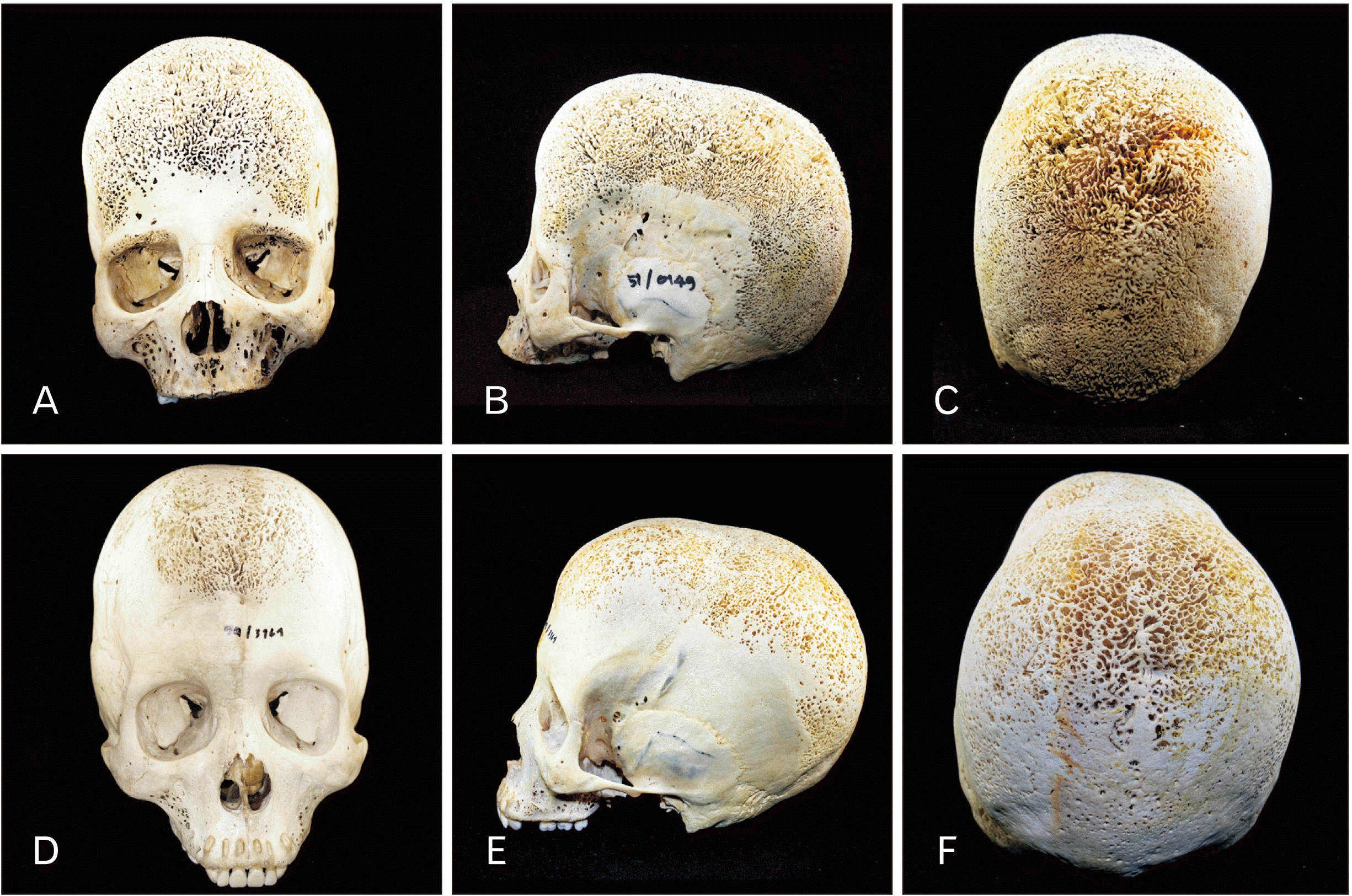
X-ray was performed for both skulls, and both showed hair-on-end appearance with and expanded diploic space (Fig. 2). Neither of the skulls showed thickening of the orbital roof when shown on X-ray (Fig. 2B, D). The distance from the outer to the inner tables ranged from 2–4 cm for both skulls, and the distance was longer in the frontal lobe than those of the parietal and occipital lobes. Complete blood counts of the two donors are presented in Table 1.
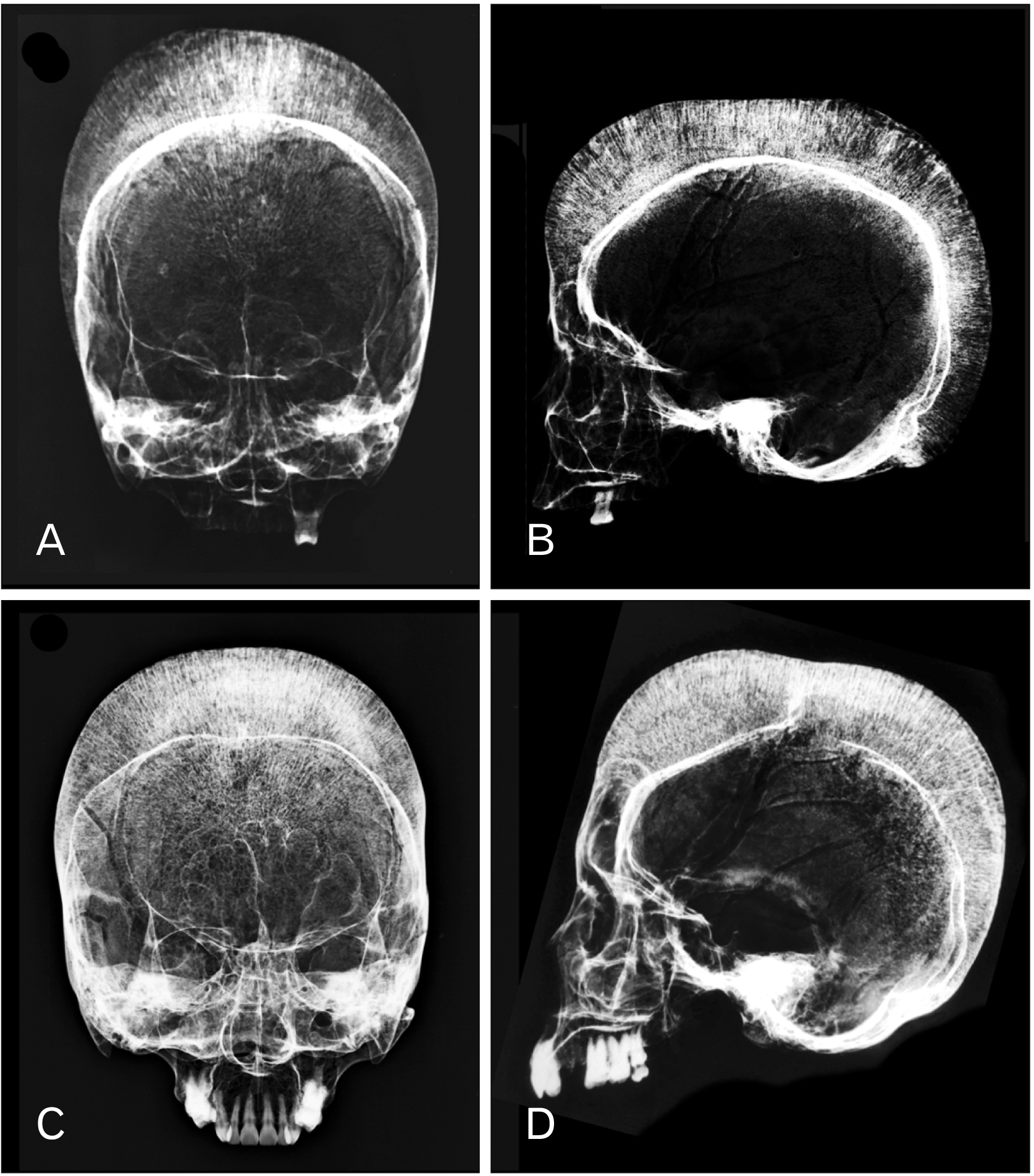 | Fig. 2X-ray frontal (A, C) and lateral view (B, D) projections of the first skull (A, B) and second skull (C, D) showing the ‘hair-on-end’ appearance.
|
Table 1
Complete blood counts of the two donors
Hb, haemoglobin; WBC, white blood cells; RBC, red blood cells; MCV, mean corpuscular volume; MCH, mean corpuscular hemoglobin; MCHC, mean cell haemoglobin concentration; RDW-CV, red cell distribution width coefficient variation; NR, not reported. Case 1 belonged to 41-year-old women diagnosed with beta thalassemia, and case 2 belonged to 35-year-old man diagnosed with unspecified type of thalassemia.
![]()
Go to :
Discussion
Cooley [5] was the first to report “peculiar bone change” in the thalassemia-related bone disease including axial and appendicular skeleton deformities. Bone modification in thalassemia is correlated with the decreased survival number of erythrocytes and decreased oxygen consumption rate in the peripheral arterial blood leading to hyperplasia of the erythroid lineage of the bone marrow [6]. Chronic hemolytic anemia in thalassemia patients causes marrow hyperplasia and hypertrophy to compensate for rapid destruction of red blood cells that exceeds the capacity of the bone marrow to produce red blood cells [7]. Marrow hyperplasia widens the medullary cavity and causes the diploic space to expand, generating the “hair-on-end” signature as seen on X-ray radiographs (Fig. 2). The increased diploic space accommodates marrow hypertrophy and hyperplasia. Ultimately, the expanding marrow causes pressure atrophy, which causes perforation and resorption of trabecular elements, producing unusually wide inter-trabecular spaces. The pressure atrophy leads to the radial arrangement of the trabecular elements, at right angles to the tables as force is directed perpendicular to the enveloping surface, to accommodate the growing marrow [8]. However, the temporal bone and occipital bone were spared and did not show any perforation and because these areas lack hemopoietic bone marrow and therefore are unaffected by marrow expansion. In addition to the skull, these changes also occur postcranially especially the spine, long bones and ribs [9, 10], because erythropoiesis takes place in these regions. The spine may lack the central squared-off depressions (H vertebra) or have vertebral body concavity (fish vertebra). There is also cortical thinning in long bones as well as cortical destruction of the ribs.
The etiology of porotic lesions of the skulls is debatable. The relationship between PH and hemolytic anemia has long been established, particularly in response to thalassemia-related anemia. Iron-deficiency as a consequence of PH was later recognized in attempt to explain the high incidence of porotic lesions in prehistoric populations, although the idea has strongly been opposed more recently [3]. Walker et al. [3] suggested high incidence of PH in prehistoric populations might be explained by the synergistic effects of malnutrition (vitamin B12 deficiency and zinc deficiency), poor sanitation, gastrointestinal infection, and cultural practices.
It is inconclusive whether the PH and CO have the same etiology. Precisely pinpointing the etiology of these porotic lesions is challenging because of other co-occurring conditions or illnesses that may have an effect on bone physiology or may induce a range of skeletal responses [11]. The independent occurrence of PH and CO, or their co-occurrence in many cases including in one of our cases, has led to different conclusions on its etiology. Some studies concluded that orbital lesions may be the initial phase before the skull is affected [12]. If CO is an earlier, less severe form of the porotic lesion that produces PH, this might explain the higher prevalence of CO [4]. However, this reasoning is denied by the fact that PH frequently occurs in skulls lacking CO [3], indicating that PH and CO are in fact non-concomitant occurrences. Lesions in different areas might be resulted from different types of anemia [13], or each type of lesion reflects separate conditions entirely [2]. It is also worth mentioning that CO could in fact be a normal developmental variant resulting from the interaction of the development of the visual system components, not necessarily a pathological response to thalassemia or anemia [4]. Only when there is convexity or thickening of the orbital roof, the CO may be deemed pathological. In the present study, the orbital roof of the first skull did not show any convexity, and therefore it is insufficient to conclude that the CO in the first skull was the result of pathological change. Also, studies on CO and PH were conducted in prehistoric populations. Their findings could be modulated by external factors such as geography, hygiene, diet and nutritional deficiency, which could have potentially overestimated the co-occurrence of CO and PH. These reasons lead to a plausible conclusion that CO cannot be considered a prerequisite for the development of PH or constant manifestation of skeletal change in thalassemia.
In summary, we have described two skulls belonging to two thalassemia patients. Both of which showed excessive porotic lesions including PH and CO. Although these two porotic lesions may be related and may co-exist in certain cases, but they are not causative of one another. The knowledge of porotic lesions of the skull in thalassemia and their radiologic appearances are of crucial importance for radiologists when encountering unusual radiologic findings.
Go to :
 XML Download
XML Download