

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Hypoxic preconditioning is the phenomenon in which short, intermittent preceding episodes of hypoxia and reperfusion confer protection to an organ/tissue from sustained ischemia/hypoxia [1]. This phenomenon is well documented to confer early as well late protection to different organs including heart, brain, kidney, and liver etc. from sustained ischemic injury [2,3]. Whole-body hypoxic preconditioning is the phenomenon in which the whole body is exposed to short, intermittent episode of ischemia and reperfusion [4], which is different from typical localized hypoxic preconditioning in which a preconditioning stimulus is delivered to a specific organ/tissue. The whole body hypoxic preconditioning offers the potential advantage of conferring multi-organ protection, which is otherwise restricted to one organ in localized preconditioning. This type of multi-organ protection may be useful in diseases in which there is a dysfunction of many organ systems such as diabetes mellitus.
Diabetes mellitus is a metabolic disorder and is characterized primarily by a disturbance in glucose metabolism. Long-standing diabetes leads to the development of a number of complications, including vascular (endothelial) dysfunction, impairment in cognitive functioning and an increase in the tendency to develop ischemia-reperfusion-induced myocardial injury [5]. The mortality rate of myocardial infarction patients suffering from diabetes mellitus is about 2–4 times higher in comparison to non-diabetic patients [6,7]. Scientists have attempted to explore the beneficial effects of hypoxic preconditioning in diabetic animals; however, the beneficial effects of hypoxic preconditioning are abolished in diabetic animals [8]. Nevertheless, there have not been studies exploring whether the loss of protection afforded by a single episode of whole body hypoxic preconditioning may be restored on repeated episodes of whole body hypoxic preconditioning in diabetic animals. In other words, whether the results of repeated episodes of hypoxic preconditioning are different from a single episode of hypoxic preconditioning in diabetic mice? Moreover, there have not been studies documenting any potential beneficial effects of repeated episodes of whole body hypoxic preconditioning on multi-organ systems in diabetic animals.
Brain-derived neurotrophic factor (BDNF) belongs to the neurotrophin family of secreted proteins and it produces biological actions through tyrosine kinase B (TrkB) receptors [9]. It was originally thought to be restricted to the brain; however, it's widespread pleiotropic effects on different organ systems are well documented [10]. In the peripheral, there are a number of potential sources of BDNF including vascular endothelium and platelets [11]. Hypoxia serves as one of the important stimuli for BDNF release [12] and different conditions which induce transient hypoxia such as exercise (producing hypoxic preconditioning) promote the release of BDNF release [3,13]. Other studies have also shown that preconditioning-induced tissue-protective effects are mediated through an increase in the expression of BDNF [14]. Moreover, studies have shown that the release of BDNF is impaired in diabetic conditions, which may be responsible for some of the diabetic complications including cognitive decline [15]. Accordingly, it may be hypothesized that diabetes-mediated attenuation of tissue-protective effects of preconditioning is mediated through a decrease in the expression of BDNF. However, there is no experimental study documenting any role of BDNF in abolishing multi-organ protection afforded by whole body hypoxic preconditioning in the diabetic state.
Nitric oxide is an endothelium-derived relaxing factor and plays a key role in hypoxic preconditioning-induced tissue protection [16]. Studies have also shown a decrease in the levels of nitric oxide in long-standing diabetes, primarily due to impairment in endothelial dysfunction [17]. It has been documented that endothelium-derived nitric oxide plays a key role in increasing the levels of BDNF [18]. Accordingly, there is a possibility of an interrelationship between nitric oxide and BDNF in diabetic-mediated attenuation of beneficial effects of hypoxic preconditioning.
Glycogen synthase kinase (GSK-3β) is a serine/threonine-protein kinase and it is a key component of the intracellular signaling cascade. An interesting point related to GSK-3β is that its enzymatic activity is decreased on its phosphorylation, which is in contrast to many other kinases [19]. Nuclear factor E2-related factor 2 (Nrf2) is a transcription factor and its key role in increasing the levels of antioxidants has been well described [20]. Studies have shown that the important contributions of GSK-3β and Nrf2 in hypoxic preconditioning-induced protection [21].
Based on these, the present study was designed to (i) explore whether repeated episodes of whole body preconditioning can confer multi-organ tissue-protective effects viz. vascular dysfunction, cognitive impairment and ischemia-reperfusion-induced enhanced myocardial injury in db/db mice (genetic model of type 2 diabetes) in comparison to a single episode of hypoxic preconditioning (ii) explore the potential role of BDNF, nitric oxide, GSK-3β and Nrf2 signaling cascade in whole body hypoxic preconditioning (single or multiple episodes)-mediated protection in db/db mice.
Go to : 
METHODS
Chemicals and animals
For the present study, male db/db mice and male C57BL/6 mice were employed. The animals were maintained on the standard diet and these had free access to drinking water. The animals were kept in the polypropylene cages in the group of three (maximum) in the animal house at a temperature of 24°–26° with relative humidity of 40%–60%. The animals were exposed to 12 h light and dark cycle. The floor of the cages was covered with husk and each cage was cleaned completely every week. The Institutional Animal Care and Use Committee of Cadre Ward 901 Hospital of the Joint Logistics Support Unit of the Chinese People's Liberation Army approved the animal ethical protocol (Ethical approval number: 202000876A). All experiments related to animals were performed as per Institutional and International ethical guidelines. The kits for estimating the levels of creatine kinase (CK-MB), lactate dehydrogenase-1 (LDH-1) and cardiac troponin T (cTnT) were purchased from Jiancheng Reagent Co, Nanjing, China. The ELISA kits were employed for the quantification of BDNF, p-GSK-3β/GSK-3β and Nrf-2 (MyBioSource, Inc., San Diego, CA, USA). ANA-12 was employed as a specific BDNF receptor antagonist (Tocris Biosciences, Minneapolis, MN, USA). The doses of L-NAME [22] and ANA-12 [23] were used as per literature reports.
Glucose levels
The mice were kept in the metabolic cages on the second last day (6th day) of 16th week for overnight fasting. Thereafter, animals were sacrificed using cervical dislocation under anesthesia (sodium thiopental) on the last day (7th day) of 16th week and therefore, the blood was isolated using cardiac puncture method. 0.5 ml of blood was centrifuged at 4,000 g at 4°C to yield plasma. Afterwards, the glucose levels were measured in the plasma using a commercially available kit based on the glucose oxidase method. Ten µl of plasma (sample) was mixed with 50 µl of reaction mixture (colorimetric probe, horse radish peroxidase and glucose oxidase) in a 96-well microtiter plate to yield a color, which OD was measured at 540 nm.
Whole-body hypoxic preconditioning
Mice were acclimatized in an environmental chamber at normal oxygen concentration for 10 min for consecutive two days before inducing hypoxic preconditioning. Hypoxic preconditioning stimulus was given by exposing the mice to four transient, alternate cycles of low O2 (8%) and normal air O2 for 10 min each. The CO2 levels were < 0.03%, humidity at 40%–50% and temperature at 22°C–24°C in the environmental chamber [4]. The C57BL/6 mice were subjected to a single episode of whole body hypoxic preconditioning on the last day of 15th week. The db/db mice were subjected to single, two or three episodes of hypoxic preconditioning. A single episode of whole body hypoxic preconditioning was given to db/db mice on the last day of 15th week. The diabetic mice were subjected to two cycles of whole body hypoxic preconditioning on the last days of 14th and 15th weeks. The diabetic mice were subjected to three episodes of whole body hypoxic preconditioning on the last days of 13th, 14th and 15th weeks.
Assessment of learning and memory in the Morris Water Maze test
In the last (16th) week of experimentation, learning and memory was assessed on five days starting from 2nd day and ending on 6th day of week. The learning and memory was assessed using the Morris Water Maze test as per standardized procedure [24,25]. The mice were subjected to five trials (each lasting 2 min) for 5 consecutive days. The first four days of trials comprised of learning and escape latency time (ELT) of day 1 was compared to ELT of day 4. The decrease in ELT signifies the learning ability. On 5th day, time spent in the target quadrant was assessed, which signifies the retrieval of learned things i.e., memory.
Vascular endothelial function
The aorta was isolated on the last day of 16th week after sacrificing mice. Thereafter, the vascular endothelial function was determined in the aortic rings, isolated from C57BL/6 and db/db mice. The arteries were cut into small rings of 2 mm length and mounted on the Myograph System. Norepinephrine was (10−5 M) was added to induce contraction in these ring segments. Thereafter, acetylcholine was added in a cumulative fashion to induce relaxation in these precontracted ring segments. The total vascular relaxation (in percentage) induced in the presence of acetylcholine was recorded for different groups. The endothelium-dependent relaxation was assessed indirectly by calculating the pIC50 value (negative log molar concentration of acetylcholine to induce 50% relaxation in precontracted arterial rings) [26].
Ischemia-reperfusion injury in isolated hearts
The hearts from non-diabetic C57BL/6 mice and db/db mice were excised on the last day of 16th week and perfused with Kreb’s Henseleit solution (118 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 1.25 mM CaCl2, 1.2 mM KH2PO4, 25 mM NaHCO3 and 11 mM glucose, pH = 7.4) on the Langendorff apparatus at 37°C. Kreb’s Henseleit was oxygenated by gassing with 95% O2/5% CO2. After stabilization of 10 min, the hearts were subjected to 30 min of global ischemia (by blocking the flow of physiological solution) and 120 min of reperfusion (by restoring the flow of physiological solution). The cardiac injury parameters viz, CK-MB, cTnT and LDH-1 were assessed in the coronary effluent, which was collected before inducing global ischemia and just after initiating reperfusion [27].
Quantification of cTnT, LDH-1, and CK-MB as biochemical markers of heart injury
The coronary effluents were collected from the isolated hearts perfused with physiological solution on the Langendorff apparatus. The samples of coronary effluents were collected before subjecting the hearts to global ischemia (before ischemia) and after reinstituting reperfusion (start of reperfusion). The levels of LDH-1, cTnT and CK-MB as specific biomarkers of heart injury were quantified in the coronary effluent using commercially available diagnostic kits. To determine the LDH-1 activity in the coronary effluent (0.1 ml), guanidine thiocyanate was added to the effluent to suppress the enzymatic activity of other isoforms of LDH including LDH-2, LDH-3, LDH-4 and LDH-5. Thereafter, lithium L-lactate and NAD+ were added in the sample (containing LDH) to yield pyruvic acid and NADH. The LDH activity was calculated by measuring the change in absorbance at 340 nm wavelength. The determination of CK-MB involved addition of ADP and creatine kinase in the coronary effluent samples (0.1 ml) to obtain ATP, which was allowed to react with glucose in the presence of hexokinase to yield glucose-6-phosphate. Thereafter, NADP+ and glucose-6-phospahte dehydrogenase were added in the reaction mixture that led to production of NADPH and its absorbance was determined at 340 nm to quantify enzymatic activity. The levels of cTnT in the coronary effluent sample (0.1 ml) were determined by sandwich assay-based ELISA kit. The wells of microplate were coated with antibody specific to cTnT (also called capture antibody). Thereafter, coronary effluent sample (containing cTnT) was added into the wells that allowed the binding of cTnT to capture antibody. It was followed by addition of biotin-conjugated antibody (detection reagent A) specific to cTnT. After washing, avidin conjugated to horseradish peroxidase (HRP) (detection reagent B) was that made complex with biotin-conjugated antibody. After washing, a substrate for HRP i.e., tetramethylbenzidine was added that yielded the color development and enzyme-substrate reaction was terminated by addition of sulphuric acid solution. The color change was measured spectrophotometrically at a wavelength of 450 nm.
Apoptosis markers
After 30 min of ischemia and 120 min of reperfusion, the heart was removed from the Langendorff apparatus. The heart was homogenized in 2 ml of phosphate buffer saline (NaCl 137 mM, KCl 2.7 mM, Na2HPO4 10 mM, KH2PO4 1.8 mM, pH = 7.4) and then, centrifuged (5,000 g for 10 min) to obtain the clear supernatant solution. Thereafter, the apoptotic markers i.e., bcl-2 and caspase 3 were measured in the supernatants of homogenized solutions. These were assessed using commercially available ELISA kits. In sandwich-based ELISA, 50 µl sample (clear supernatant) was added in the well, which was followed by addition of 50 µl of antibody cocktail to all wells. After incubation of 1 h, wells were washed with wash buffer followed by addition of 100 µl tetramethylbenzidine to yield a color, whose OD was measured at 450 nm. The total protein content in the homogenate solution was measured by Lowery's method in which 100 µl of sample was mixed with 1 ml of color complex forming compound (sodium potassium tartrate, copper sulfate, Folin-Ciocalteau reagent) to yield a color, whose OD was measured at 750 nm. The apoptotic markers (bcl-2 and caspase 3) were represented with respect to total protein content [28,29].
Quantification of serum nitrite, BDNF, p-GSK-3β/GSK-3β ratio, Nrf-2 levels, and acetylcholinesterase activity
The blood (0.2 ml) was isolated from retroorbital sinus of mice on the last day of 15th week, 1 h after the completion of hypoxic preconditioning protocol. The blood was left undisturbed for about 15–30 min to clot at room temperature. Thereafter, it was centrifuged at 2,000 g for 10 min to separate the serum. The levels of nitrite and BDNF were quantified in the serum of C57BL/6 and db/db mice. The determination of nitrite in the serum sample levels was done by spectrophotometric method [30]. In this test, the nitric oxide assay kit was employed to measure the total nitrite levels that included the measurement of nitrite present in sample (direct measurement) as well as nitrite derived from nitrates (nitrate reductase converted nitrate to nitrite). Fifty µl of sample was allowed to react with Greiss reagent to give azo dye, which OD was measured at 540 nm.
The quantitative assessment of p-GSK-3β/GSK-3β ratio and Nrf2 levels in the supernatants of heart homogenate solution and BDNF in the serum was done using commercially available ELISA kits, based on the Sandwich assay principle. The clear supernatant solution (10 µl, for GSK-3β ratio and Nrf2), or serum (10 µl for BDNF) was added into the 96 welled microtiter plates, whose wells were coated with capture antibodies. Thereafter, the detection reagent A (containing the biotinylated, detection antibodies) was added and after washing, detection reagent B (containing streptavidin-HRP conjugate) was added, which made the complex with biotin attached to detection antibody. After washing, a substrate for HRP (tetramethylbenzidine) was added to obtain the color, whose absorbance was measured at 450 nm.
Along with heart and aorta, the brains were also isolated from sacrificed mice. The brain was homogenized in 2 ml of phosphate buffer saline (pH = 7.4) and centrifuged (5,000 g for 10 min) to obtain the clear supernatant solution. The acetylcholinesterase activity was assessed in the clear supernatant solution of the brain homogenates using a colorimetric method [31,32]. 0.5 µl of sample was added in 96 welled microtitre plate and volume was made up to 50 µl by the addition of assay buffer, which was followed by addition of 50 µl of AChE reaction mixture. After incubation for 15 min, the absorbance was noted at 410 nm.
Experimental design
The following experimental groups were employed in the present study and each group comprised of eight animals. All experiments (behavioral, biochemical, physiological) were conducted on eight animals in each experimental group.
i. Control: The hearts from non-diabetic, non-ischemic mice were homogenized for the measurement of biochemical parameters including p-GSK-3β/GSK-3β ratio and Nrf-2 levels.
ii. Normal: C57BL/6 (non-diabetic) mice were kept for 16 weeks. In the last five days of the 16th week, cognitive function was assessed using the Morris Water Maze test as described in section 2.4. On the last day of the 16th week, mice were sacrificed to isolate the blood (for glucose determination), hearts, brains (for the measurement of acetylcholinesterase activity) and aortic rings. The heart was subjected to ischemia-reperfusion on the Langendorff apparatus and aortic rings were employed to assess vascular functionality. The biochemical markers of injury (LDH-1, CK-MB, cTnT) were measured in coronary effluent. The assessment of apoptotic markers (Bcl-2 and caspase 3), GSK-3β/GSK-3β ratio and Nrf-2 levels was done in the supernatants of heart homogenate solutions. The brains were homogenized for the quantification of acetylcholinesterase activity.
iii. Diabetes: db/db mice were kept for 16 weeks and the same protocol was followed as described in group ii.
iv. Single-cycle of whole body hypoxic preconditioning in normal: C57BL/6 mice were subjected to a single cycle of whole body hypoxic preconditioning (section 2.3) on the last day of 15th week. After one hour of preconditioning stimulus, the serum nitrite and serum BDNF levels were measured. The rest of the protocol was the same as described in group ii.
v. Single-cycle of whole body hypoxic preconditioning in diabetes: The same protocol was followed in db/db mice as described in group iv.
vi. Two cycles of whole body hypoxic preconditioning in diabetes: db/db mice were subjected to two cycles of whole body hypoxic preconditioning on the last days of 14th and 15th weeks. The rest of the protocol was the same as in group iv.
vii. Three cycles of whole body hypoxic preconditioning in diabetes: db/db mice were subjected to three cycles of whole body hypoxic preconditioning on the lasts day of 13th, 14th and 15th weeks. The rest of the protocol was the same as in group iv.
viii. ANA-12 TrkB antagonist (0.25 mg/kg) in one cycle of whole body hypoxic preconditioning in normal: ANA-12 (0.25 mg/kg i.p.) was given to C57BL/6 mice before subjecting to whole body hypoxic preconditioning. The rest of the protocol was the same as in group iv.
ix. ANA-12 TrkB antagonist (0.50 mg/kg) in one cycle of whole body hypoxic preconditioning in normal: ANA-12 (0.50 mg/kg i.p.) was given to C57BL/6 mice before subjecting to whole body hypoxic preconditioning. The rest of the protocol was the same as in group iv.
x. ANA-12 TrkB antagonist (0.50 mg/kg) in three cycles of whole body hypoxic preconditioning in diabetes: ANA-12 (0.50 mg/kg i.p.) was given to db/db mice before the last episode of whole body hypoxic preconditioning (i.e. at the end of 15th week). The rest of the protocol was the same as in group iv.
xi. L-NAME (15 mg/kg) in one cycle of whole body hypoxic preconditioning in normal: L-NAME (15 mg/kg i.p.) was given to C57BL/6 mice before subjecting to whole body hypoxic preconditioning. The rest of the protocol was the same as in group iv.
xii. L-NAME (30 mg/kg) in one cycle of whole body hypoxic preconditioning in normal: L-NAME (30 mg/kg i.p.) was given to C57BL/6 mice before subjecting to whole body hypoxic preconditioning. The rest of the protocol was the same as in group iv.
xiii. L-NAME (30 mg/kg) in three cycles of whole body hypoxic preconditioning in diabetes: L-NAME (30 mg/kg i.p.) was given to db/db mice before last episode of whole body hypoxic preconditioning (i.e., at the end of 15th week). The rest of the protocol was the same as in group iv.
Statistical analysis
The data of the study were presented as mean ± standard deviation. The data of CK-MB, cTnT, LDH-1, ELT, vascular functionality were statistically analyzed using Two Way Repeated Measure ANOVA. The data of other parameters were compared using One-Way ANOVA. Tukey’s post-hoc test was employed for multiple comparisons. p < 0.05 was considered to be statistically significant.
Go to :
RESULTS
Long standing diabetic condition worsened ischemia-reperfusion-induced myocardial injury in isolated hearts
There was a significant increase in myocardial injury in hearts isolated from non-diabetic, C57BL/6 mice in response to 30 min of ischemia and 120 min of reperfusion as assessed by a marked increase in the levels of CK-MB (Fig. 1), cTnT (Fig. 2) and LDH-1 (Fig. 3) in the coronary effluent. There was a marked increase in the levels of cardiac injury parameters during the reperfusion phase in comparison to basal state i.e. before subjecting to ischemia. The extent of release of these biochemical parameters of cardiac injury i.e., CK-MB, cTnT and LDH-1 from the hearts isolated from db/db mice was even more significant in response to 30 min of ischemia and 120 min of reperfusion. There was a significant increase in the apoptotic markers (bcl-2 and caspase 3) in non-diabetic hearts subjected to an ischemia-reperfusion injury (Fig. 4). However, this rise was more significant in db/db hearts in comparison to C57BL/6 mice clearly indicating that long-standing diabetes mellitus significantly enhances ischemia-reperfusion-induced myocardial injury.
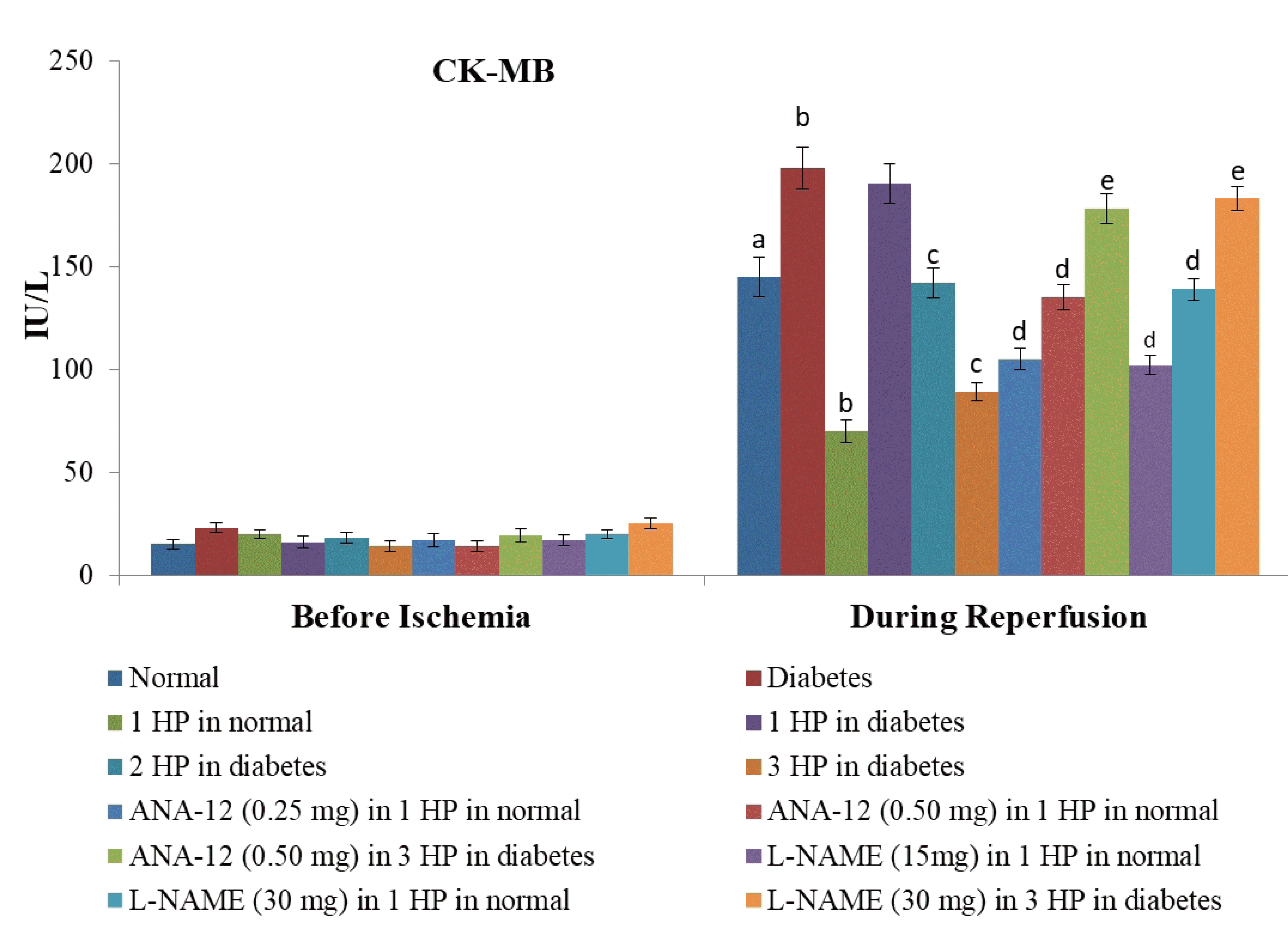 | Fig. 1Effects of different interventions on the extent of myocardial injury assessed by measuring the release of specific marker of heart injury i.e., creatine kinase (CK)-MB in the coronary effluent.The measurement was done before subjecting the heart to ischemia (before ischemia) and at the start of reperfusion. HP, hypoxic preconditioning; 1 HP, single episode of hypoxic preconditioning. ap < 0.05 vs. basal; bp < 0.05 vs. normal; cp < 0.05 vs. diabetes; dp < 0.05 vs. 1 HP in normal; ep < 0.05 vs. 3 HP in diabetes.
|
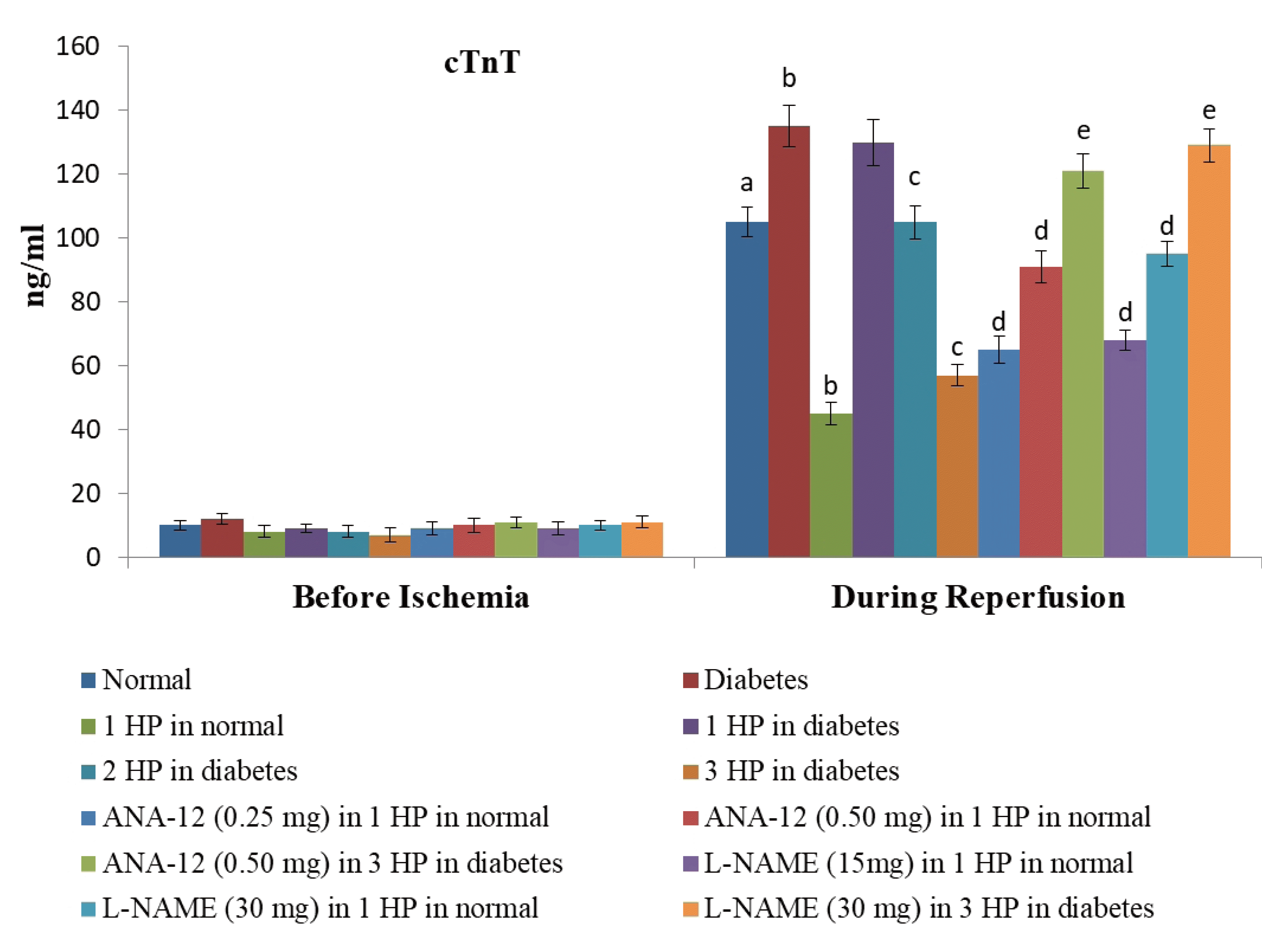 | Fig. 2Effects of different interventions on the extent of myocardial injury assessed by measuring the release of specific marker of heart injury i.e., cardiac troponin T (cTnT) in the coronary effluent.The measurement was done before subjecting the heart to ischemia (before ischemia) and at the start of reperfusion. HP, hypoxic preconditioning; 1 HP, single episode of hypoxic preconditioning. ap < 0.05 vs. basal; bp < 0.05 vs. normal; cp < 0.05 vs. diabetes; dp < 0.05 vs. 1 HP in normal; ep < 0.05 vs. 3 HP in diabetes.
|
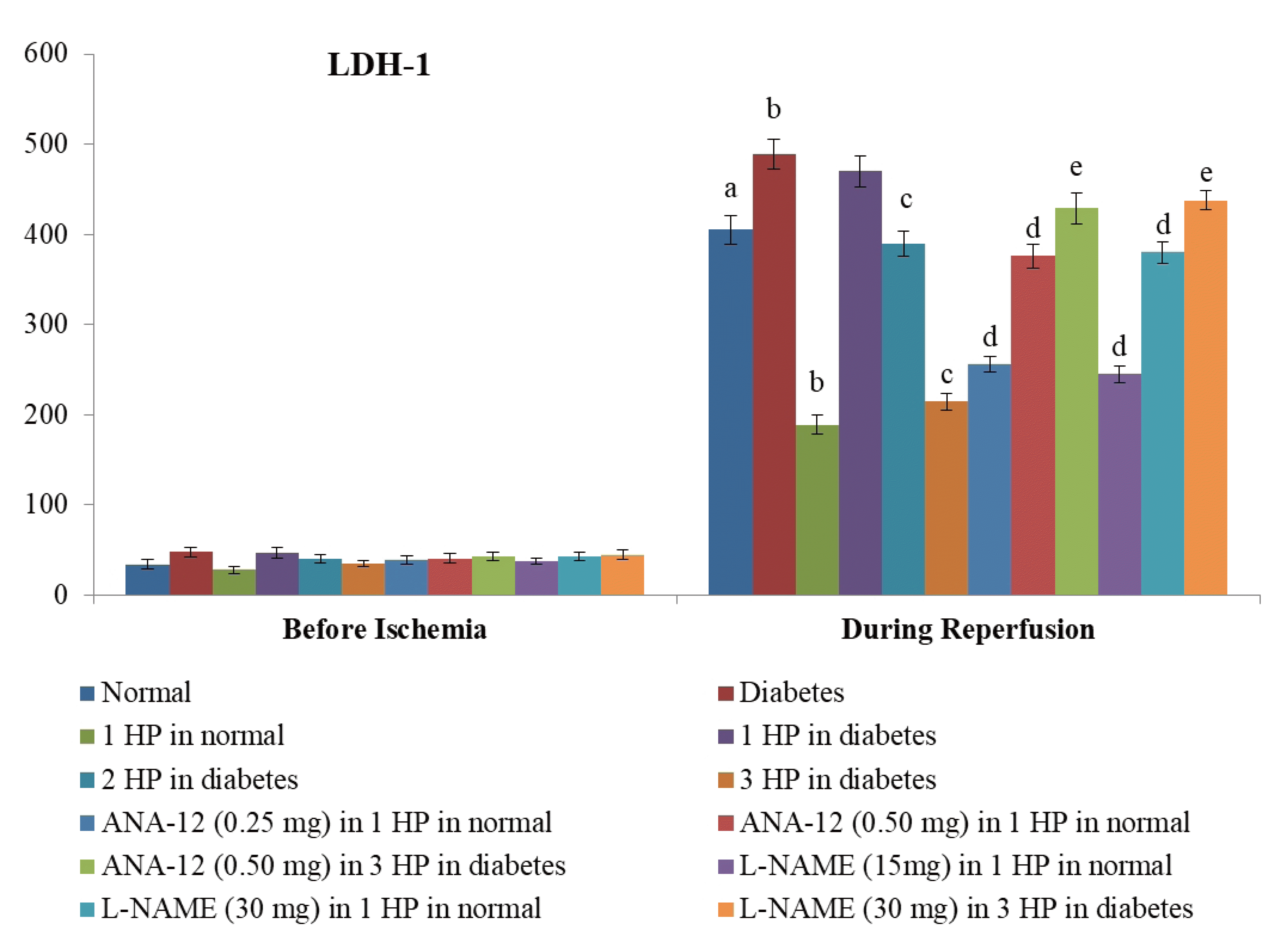 | Fig. 3Effects of different interventions on the extent of myocardial injury assessed by measuring the release of specific marker of heart injury i.e., lactate dehydrogenase-1 (LDH-1) in the coronary effluent.The measurement was done before subjecting the heart to ischemia (before ischemia) and at the start of reperfusion. HP, hypoxic preconditioning; 1 HP, single episode of hypoxic preconditioning. ap < 0.05 vs. basal; bp < 0.05 vs. normal; cp < 0.05 vs. diabetes; dp < 0.05 vs. 1 HP in normal; ep < 0.05 vs. 3 HP in diabetes.
|
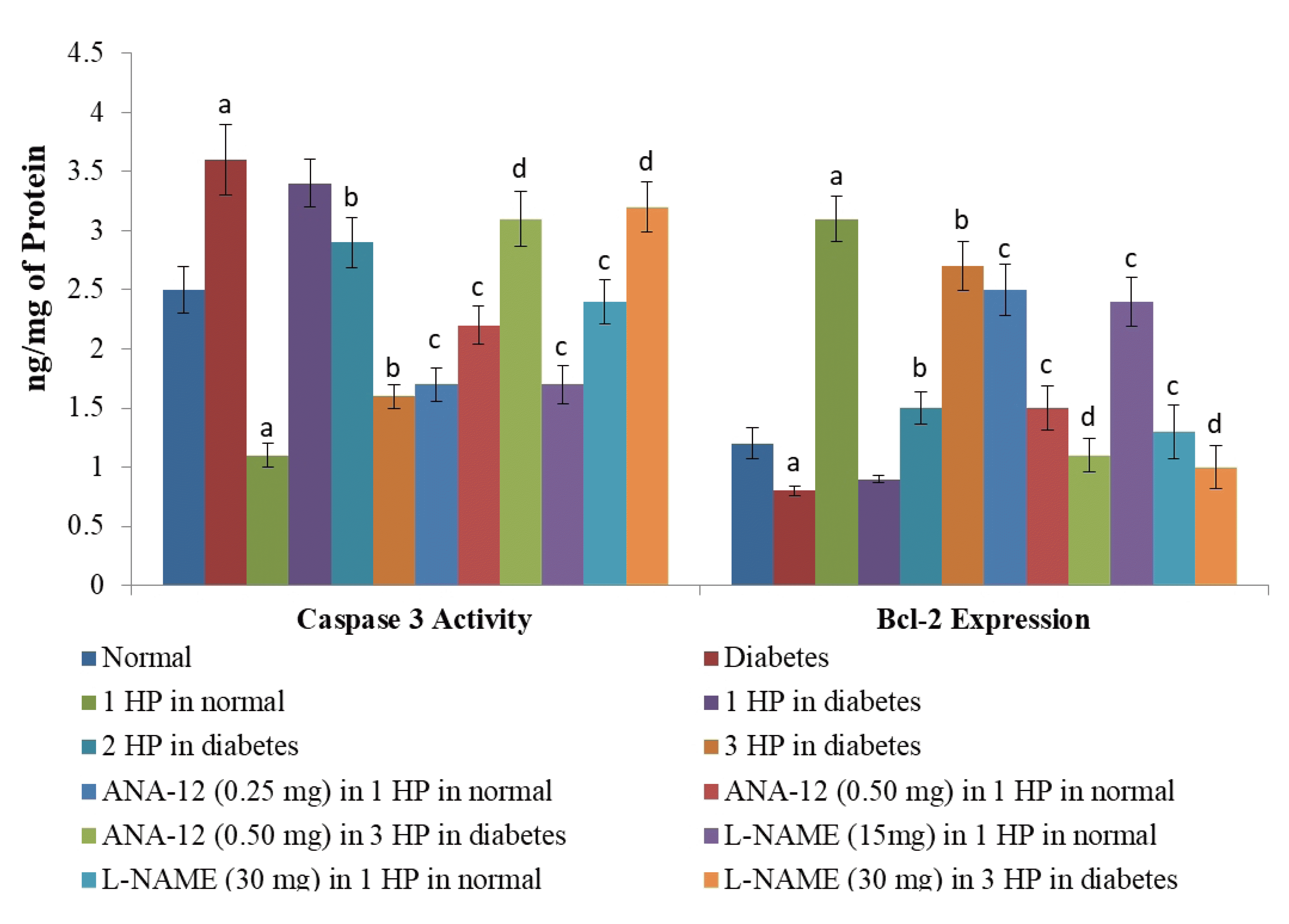 | Fig. 4Effects of different interventions on the markers of apoptosis i.e., caspase 3 activity and Bcl-2 expression in the heart homogenates after ischemia-reperfusion injury.HP, hypoxic preconditioning; 1 HP, single episode of hypoxic preconditioning. ap < 0.05 vs. normal; bp < 0.05 vs. diabetes; cp < 0.05 vs. 1 HP in normal; dp < 0.05 vs. 3 HP in diabetes.
|
Whole-body hypoxic preconditioning protected hearts from ischemia-reperfusion injury
A single episode of whole body preconditioning (at the end of 15th week) protected hearts of C57BL/6 mice from the deleterious effects of ischemia-reperfusion injury and there was significant reduction in the levels of CK-MB (Fig. 1), cTnT (Fig. 2) and LDH-1 (Fig. 3) in coronary effluent and apoptotic markers (caspase 3 and bcl-2) in heart homogenates (Fig. 4). A single exposure to preconditioning (at the end of 15th week) failed to induce cardioprotection in db/db mice. Nevertheless, repeated exposures (two and three) to preconditioning stimulus triggered protection in the hearts of diabetic mice against ischemia-reperfusion injury. The extent of protection imparted by three episodes of preconditioning (at the end of 13th, 14th and 15th week) was significantly higher than two episodes of preconditioning (at the end of 14th and 15th week).
Repeated episodes of whole body hypoxic preconditioning preserves decline in cognitive functions in diabetic mice
On a Morris water maze test, there was a significant decline in the 4th day ELT in comparison to the 1st day ELT in C57BL/6 mice (assessed during the first four days of acquisition trials) indicating the normal learning ability of non-diabetic animals. On the other hand, no such significant decline in 4th day ELT was noted in db/db mice indicating the impaired learning in long-standing diabetic mice. Moreover, time spent in the target quadrant (as retrieval index) assessed on the 5th day of trial was significantly higher in C57BL/6 mice in comparison to db/db mice, again suggesting the impairment in memory in the long-standing diabetic state. Exposure to three episodes of whole body hypoxic preconditioning in db/db mice led to a significant decline in 4th day ELT and an increase in time spent in the target quadrant on the 5th day in diabetic mice suggesting the improvement in cognitive functions in diabetic mice. A single episode of preconditioning did not modulate ELT (Table 1) and time spent in the target quadrant in C57BL/6 and db/db mice (Fig. 5).
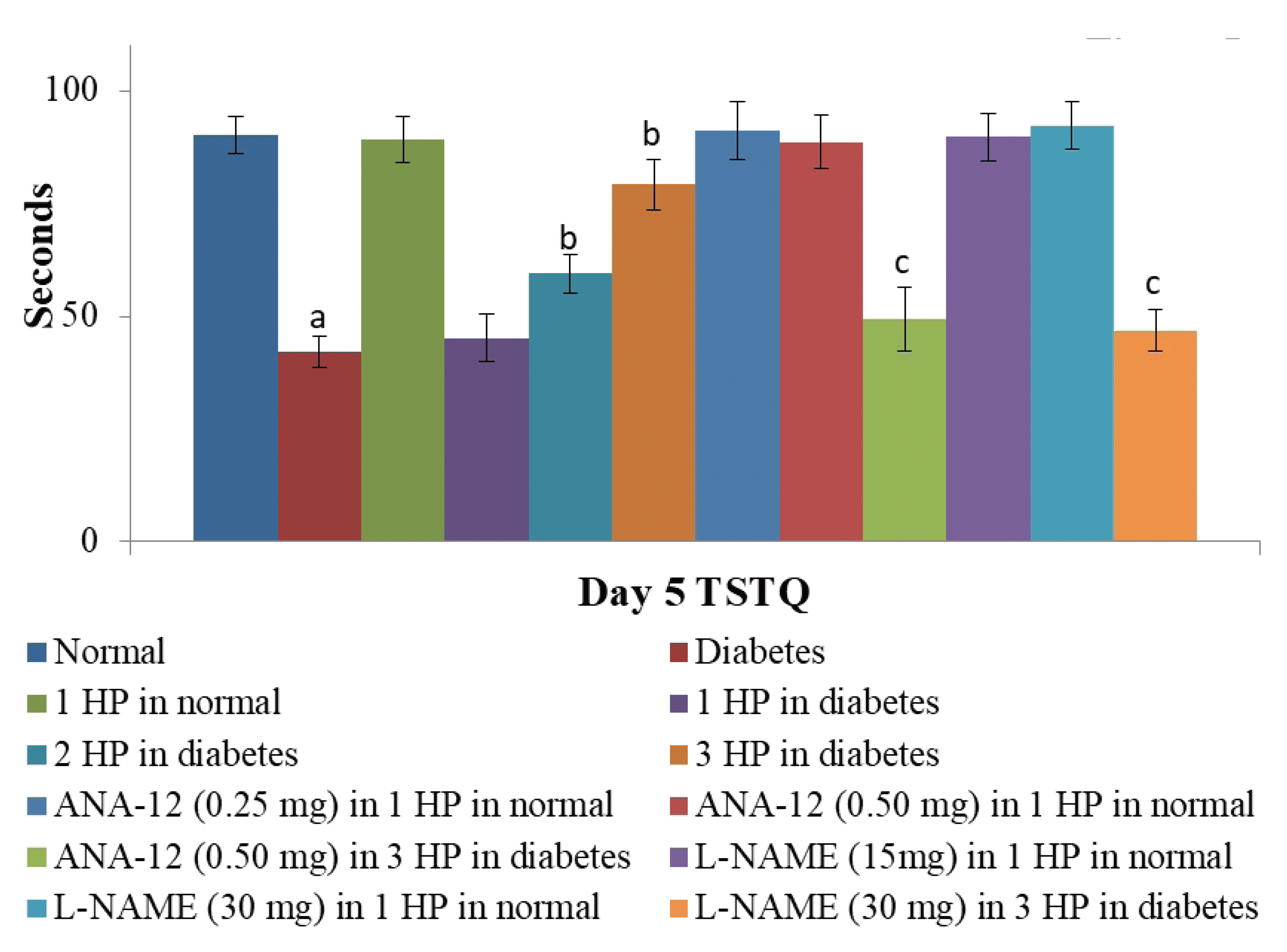 | Fig. 5Effects of different interventions on the memory retrieval (how much information is retained following learning) in the form of time spent in target quadrant (TSTQ).It was assessed on the 5th day of trial in the Morris Water Maze test. HP, hypoxic preconditioning; 1 HP, single episode of hypoxic preconditioning. ap < 0.05 vs. normal; bp < 0.05 vs. diabetes; cp < 0.05 vs. 3 HP in diabetes.
|
Table 1
Effects of different interventions on the learning ability of mice by measuring the escape latency time (ELT) from day 1 to 4 in the Morris Water Maze test
| S. No | Groups | Day 1 ELT (sec) | Day 4 ELT (sec) |
|---|---|---|---|
| 1 | Normal | 90.2 ± 5.7 | 35.6 ± 3.2a |
| 2 | Diabetes | 94.4 ± 6.2 | 80.1 ± 5.2b |
| 3 | Single cycle of whole body hypoxic preconditioning in normal | 89.1 ± 4.8 | 33.2 ± 4.1 |
| 4 | Single cycle of whole body hypoxic preconditioning in diabetes | 93.1 ± 6.1 | 76.3 ± 5.3 |
| 5 | Two cycles of whole body hypoxic preconditioning in diabetes | 92.1 ± 5.8 | 67.2 ± 6.1c |
| 6 | Three cycles of whole body hypoxic preconditioning in diabetes | 91.3 ± 4.3 | 49.3 ± 3.9c |
| 7 | ANA-12 TrkB antagonist (0.25 mg/kg) in one cycle of whole body hypoxic preconditioning in normal | 91.3 ± 4.3 | 37.2 ± 3.9 |
| 8 | ANA-12 TrkB antagonist (0.50 mg/kg) in one cycle of whole body hypoxic preconditioning in normal | 91.9 ± 3.1 | 38.1 ± 4.1 |
| 9 | ANA-12 TrkB antagonist (0.50 mg/kg) in three cycles of whole body hypoxic preconditioning in diabetes | 93.4 ± 7.1 | 72. 9 ± 6.6d |
| 10 | L-NAME (15 mg/kg) in one cycle of whole body hypoxic preconditioning in normal | 90.5 ± 6.2 | 33.4 ± 3.7 |
| 11 | L-NAME (30 mg/kg) in one cycle of whole body hypoxic preconditioning in normal | 91.6 ± 4.8 | 34.2 ± 3.1 |
| 12 | L-NAME (30 mg/kg) in three cycles of whole body hypoxic preconditioning in diabetes | 95.3 ± 6.1 | 73.5 ± 5.4d |
![]()
Repeated episodes of whole body hypoxic preconditioning preserve vascular functionality in diabetic mice
In C57BL/6 mice, the cumulative addition of acetylcholine led to nearly complete relaxation of norepinephrine-precontracted aortic rings suggesting the normal functioning of vascular endothelium in non-diabetic mice. However, there was impairment in vascular functionality in db/db mice as the cumulative addition of acetylcholine failed to relax norepinephrine-precontracted aortic rings in a complete manner (Table 2). Accordingly, the pIC50 values were significantly higher in db/db mice as compared to C57BL/6 mice (Table 3). Repeated exposure to three episodes of whole body hypoxic preconditioning led to significant improvement in vascular relaxation and a decrease in pIC50 value in db/db mice suggesting the improvement in vascular functionality in diabetic mice. A single episode of preconditioning did not modulate vascular relaxation and decrease in pIC50 value in C57BL/6 and db/db mice.
Table 2
Effects of different interventions on the endothelial function was assessed by measuring the vascular relaxation in norepinephrine-precontracted aortic rings on cumulative addition of acetylcholine
| S. No | Groups | Percent contraction on cumulative addition of acetylcholine (Log molar concentration) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| –9.0 | –8.5 | –8.0 | –7.5 | –7.0 | –6.5 | –6.0 | –5.5 | –5.0 | ||
| 1 | Normal | 95.1 ± 2.1 | 87.2 ± 2.4 | 74.3 ± 2.0 | 61.1 ± 2.3 | 49.1 ± 2.6 | 31.1 ± 2.1 | 18.1 ± 1.8 | 9.1 ± 1.1 | 2.1 ± 0.3 |
| 2 | Diabetes | 99.1 ± 1.6 | 94.2 ± 1.3a | 89.3 ± 2.3a | 81.1 ± 2.0a | 74.1 ± 2.3a | 69.1 ± 2.3a | 61.0 ± 2.6a | 54.1 ± 2.1a | 47.4 ± 3.3a |
| 3 | 1 cycle of preconditioning in normal | 97.1 ± 2.6 | 88.2 ± 2.2 | 74.1 ± 2.7 | 60.1 ± 2.1 | 49.9 ± 2.0 | 32.1 ± 2.7 | 20.1 ± 1.5 | 10.1 ± 1.2 | 2.7 ± 0.4 |
| 4 | 1 cycle of preconditioning in diabetes | 98.1 ± 1.5 | 92.1 ± 1.8 | 87.2 ± 1.6 | 80.1 ± 1.3 | 72.4 ± 2.0 | 67.4 ± 2.4 | 58.2 ± 1.6 | 53.5 ± 2.4 | 45.4 ± 2.1 |
| 5 | 2 cycles of preconditioning in diabetes | 97.1 ± 2.0 | 91.4 ± 2.0b | 84.2 ± 2.7b | 77.1 ± 2.4b | 69.4 ± 2.5b | 61.3 ± 2.0b | 54.4 ± 2.1b | 49.3 ± 2.4b | 40.4 ± 2.3b |
| 6 | 3 cycles of preconditioning in diabetes | 96.1 ± 2.4 | 89.4 ± 2.7b | 79.5 ± 2.2b | 67.1 ± 2.2b | 59.3 ± 2.0b | 48.3 ± 2.4b | 31.3 ± 2.4b | 22.1 ± 1.4b | 11.4 ± 1.1b |
| 7 | ANA-12 (0.25 mg/kg) in 1 cycle of preconditioning in normal | 94.2 ± 2.0 | 86.4 ± 2.5 | 72.5 ± 2.3 | 59.3 ± 2.5 | 47.3 ± 3.4 | 30.1 ± 1.9 | 17.4 ± 1.2 | 8.4 ± 0.8 | 1.9 ± 0.3 |
| 8 | ANA-12 (0.50 mg/kg) in 1 cycle of preconditioning in normal | 93.3 ± 2.8 | 86.1 ± 2.4 | 73.5 ± 2.4 | 61.4 ± 2.5 | 48.4 ± 2.3 | 33.5 ± 2.4 | 21.6 ± 1.7 | 12.5 ± 1.4 | 2.9 ± 0.5 |
| 9 | ANA-12 (0.50 mg/kg) in 3 cycles of preconditioning in diabetes | 98.1 ± 1.9 | 91.3 ± 2.0 | 87.9 ± 2.1c | 78.5 ± 2.4c | 70.4 ± 2.5c | 65.1 ± 2.1c | 59.4 ± 2.1c | 52.1 ± 2.4c | 45.4 ± 1.7c |
| 10 | L-NAME (15 mg/kg) in 1 cycle of preconditioning in normal | 95.1 ± 2.3 | 89.1 ± 2.4 | 76.4 ± 2.2 | 61.8 ± 2.4 | 49.1 ± 2.5 | 31.1 ± 2.2 | 22.1 ± 1.4 | 11.4 ± 1.4 | 3.7 ± 0.5 |
| 11 | L-NAME (30 mg/kg) in 1 cycle of preconditioning in normal | 94.9 ± 2.1 | 87.3 ± 2.8 | 72.4 ± 2.3 | 60.6 ± 2.5 | 48.1 ± 2.4 | 30.5 ± 2.5 | 18.1 ± 1.9 | 7.1 ± 0.4 | 1.9 ± 0.2 |
| 12 | L-NAME (30 mg/kg) in 3 cycles of preconditioning in diabetes | 98.9 ± 2.3 | 92.1 ± 2.1 | 88.0 ± 1.9c | 80.5 ± 2.1c | 71.3 ± 2.7c | 67.3 ± 1.7c | 61.4 ± 1.8c | 54.5 ± 1.3c | 46.6 ± 1.1c |
![]()
Table 3
Effects of different interventions on pIC50 values i.e., negative log molar concentration of acetylcholine that produced 50% relaxation in norepinephrine-precontracted aortic rings
| S. No | Groups | pIC50 |
|---|---|---|
| 1 | Normal | 7.13 ± 1.30 |
| 2 | Diabetes | 5.21 ± 1.41a |
| 3 | Single cycle of whole body hypoxic preconditioning in normal | 7.12 ± 1.23 |
| 4 | Single cycle of whole body hypoxic preconditioning in diabetes | 5.24 ± 1.31 |
| 5 | Two cycles of whole body hypoxic preconditioning in diabetes | 5.83 ± 1.44 |
| 6 | Three cycles of whole body hypoxic preconditioning in diabetes | 6.41 ± 1.28b |
| 7 | ANA-12 TrkB antagonist (0.25 mg/kg) in one cycle of whole body hypoxic preconditioning in normal | 7.09 ± 1.36 |
| 8 | ANA-12 TrkB antagonist (0.50 mg/kg) in one cycle of whole body hypoxic preconditioning in normal | 7.12 ± 1.28 |
| 9 | ANA-12 TrkB antagonist (0.50 mg/kg) in three cycles of whole body hypoxic preconditioning in diabetes | 5.25 ± 1.31c |
| 10 | L-NAME (15 mg/kg) in one cycle of whole body hypoxic preconditioning in normal | 7.15 ± 1.24 |
| 11 | L-NAME (30 mg/kg) in one cycle of whole body hypoxic preconditioning in normal | 7.10 ± 1.24 |
| 12 | L-NAME (30 mg/kg) in three cycles of whole body hypoxic preconditioning in diabetes | 5.12 ± 1.39c |
![]()
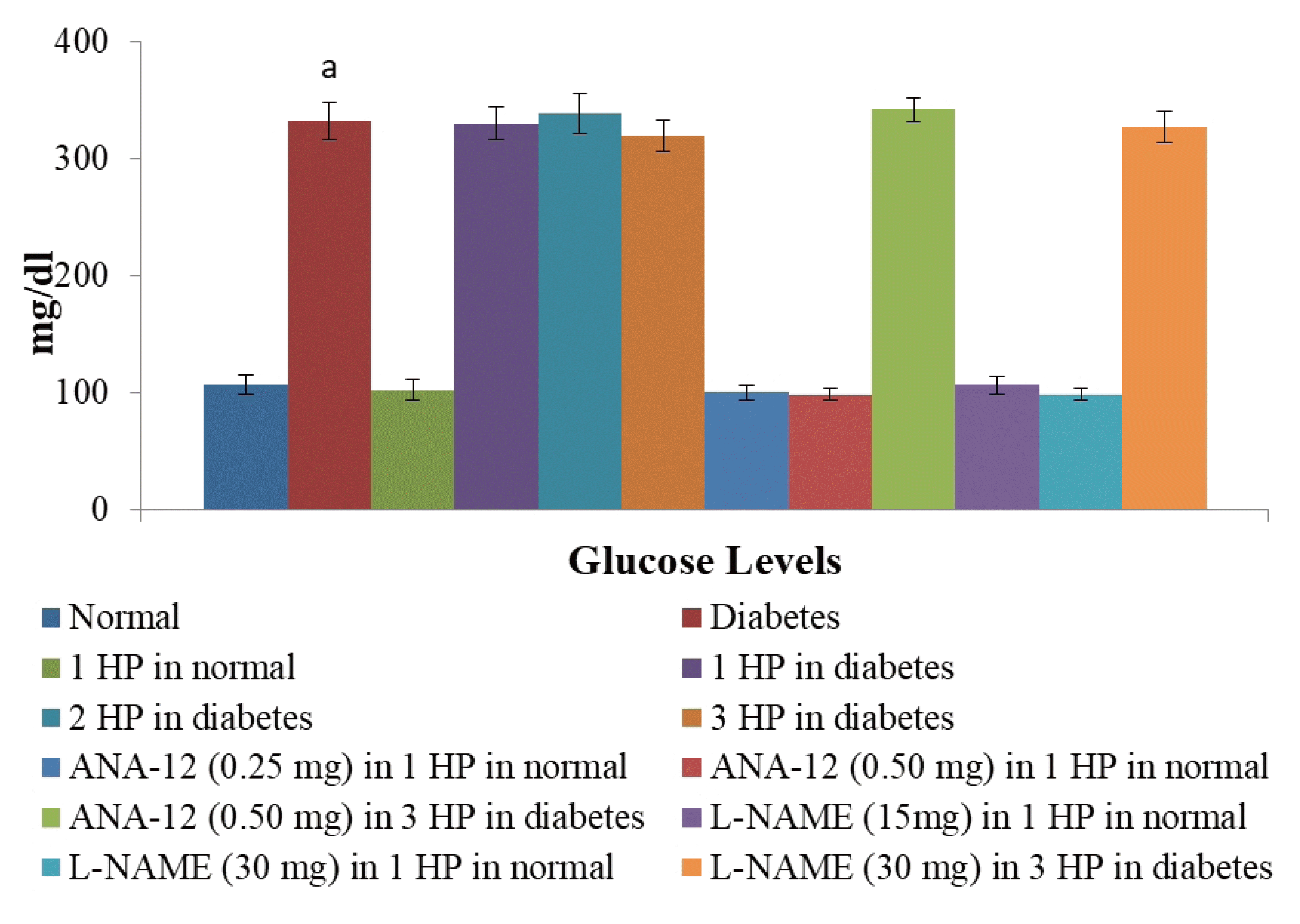
Repeated episodes of whole body hypoxic preconditioning did not modulate the blood sugar levels
There was a significant increase in the blood sugar levels in db/db mice as assessed at the end of the 16th week in comparison to C57BL/6 mice. Single or repeated episodes of whole body hypoxic preconditioning did not modulate the blood sugar levels in db/db or C57BL/6 mice in a significant manner (Fig. 6).
Whole-body preconditioning significantly influences the biochemical parameters in db/db and C57BL/6 mice
There was a significant increase in the levels of serum nitrite levels (an indirect measure of nitric oxide) (Fig. 7) and serum BDNF levels (Fig. 8) in C57BL/6 mice in response to a single episode of whole body hypoxic preconditioning. However, a similar rise in the serum nitrite and serum BDNF levels was observed in db/db mice after three episodes of preconditioning. Moreover, there was a significant decrease in the p-GSK-3β/GSK-3β ratio and Nrf-2 levels (Fig. 9) in the supernatants of heart homogenates of C57BL/6 and db/db mice following an ischemia-reperfusion injury. A single episode of preconditioning in C57BL/6 and three episodes of preconditioning in db/db mice led to the significant restoration of p-GSK-3β/GSK-3β ratio and Nrf-2 levels in ischemia-reperfusion-subjected hearts. There was a significant increase in brain acetylcholinesterase activity in db/db mice in comparison to C57BL/6 mice, which was abolished in response to three episodes of preconditioning. There was no influence of preconditioning on the brain acetylcholinesterase activity in C57BL/6 mice (Table 4).
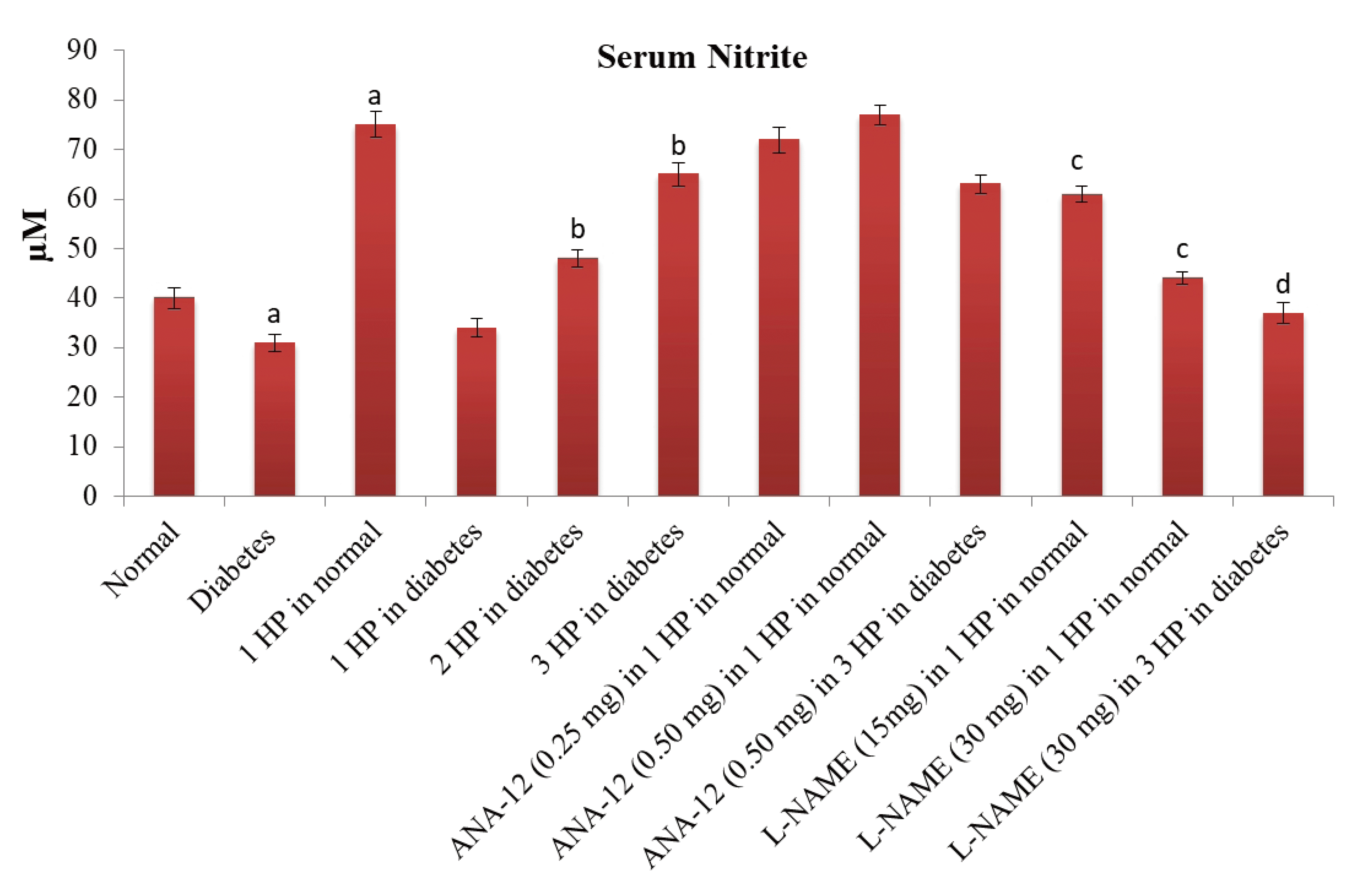 | Fig. 7Effects of different interventions on the nitric oxide, which was indirectly assessed by measuring the levels of nitrite in the serum.The levels of serum nitrite were measured 1 h after hypoxic preconditioning stimulus. HP, hypoxic preconditioning; 1 HP, single episode of hypoxic preconditioning. ap < 0.05 vs. normal; bp < 0.05 vs. diabetes; cp < 0.05 vs. 1 HP in normal; dp < 0.05 vs. 3 HP in diabetes.
|
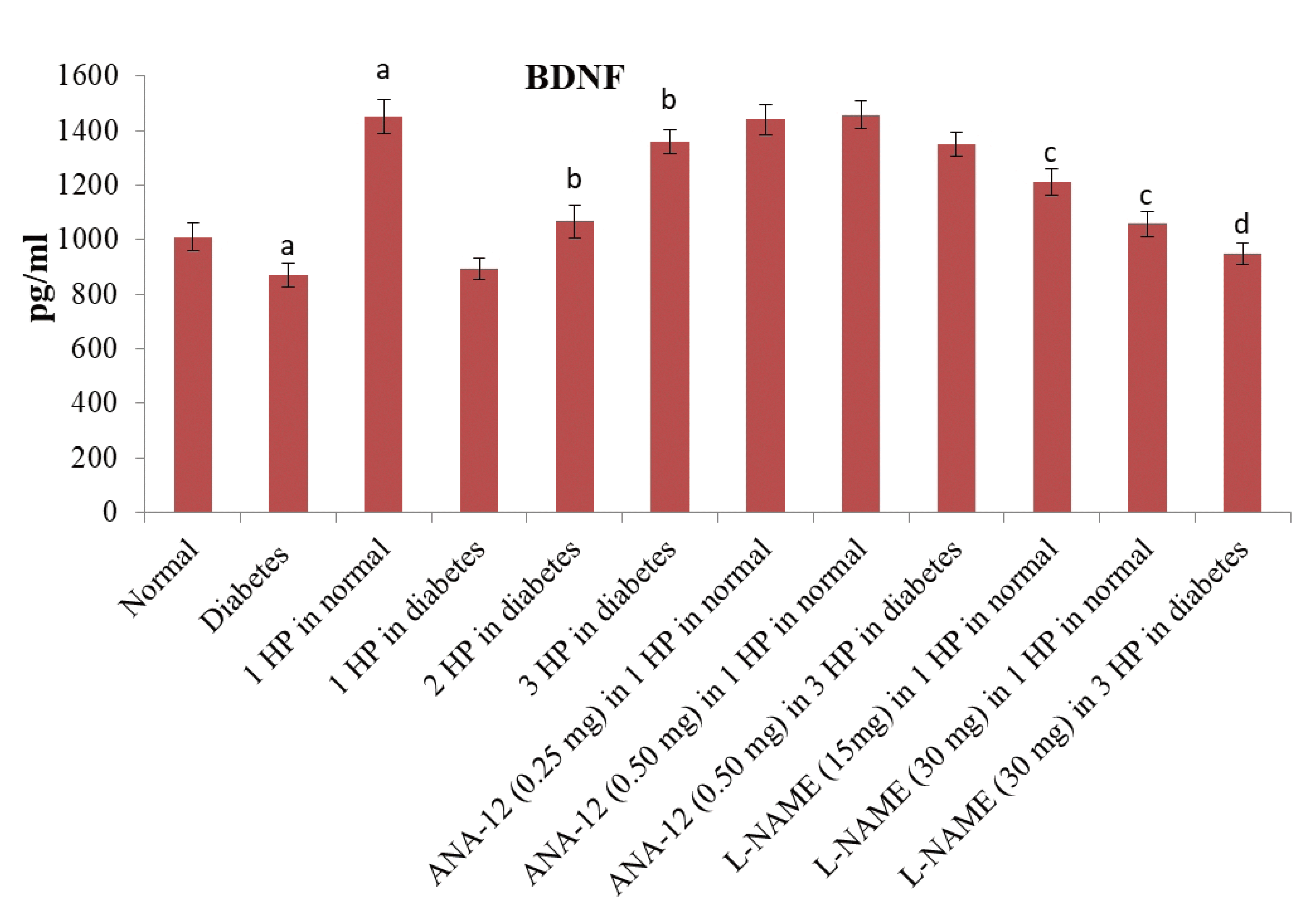 | Fig. 8Effects of different interventions on the pharmacological target employed in this study i.e., brain-derived neurotrophic factor (BDNF).The levels of BDNF were measured in the serum 1 h after hypoxic preconditioning stimulus. HP, hypoxic preconditioning; 1 HP, single episode of hypoxic preconditioning. ap < 0.05 vs. normal; bp < 0.05 vs. diabetes; cp < 0.05 vs. 1 HP in normal; dp < 0.05 vs. 3 HP in diabetes.
|
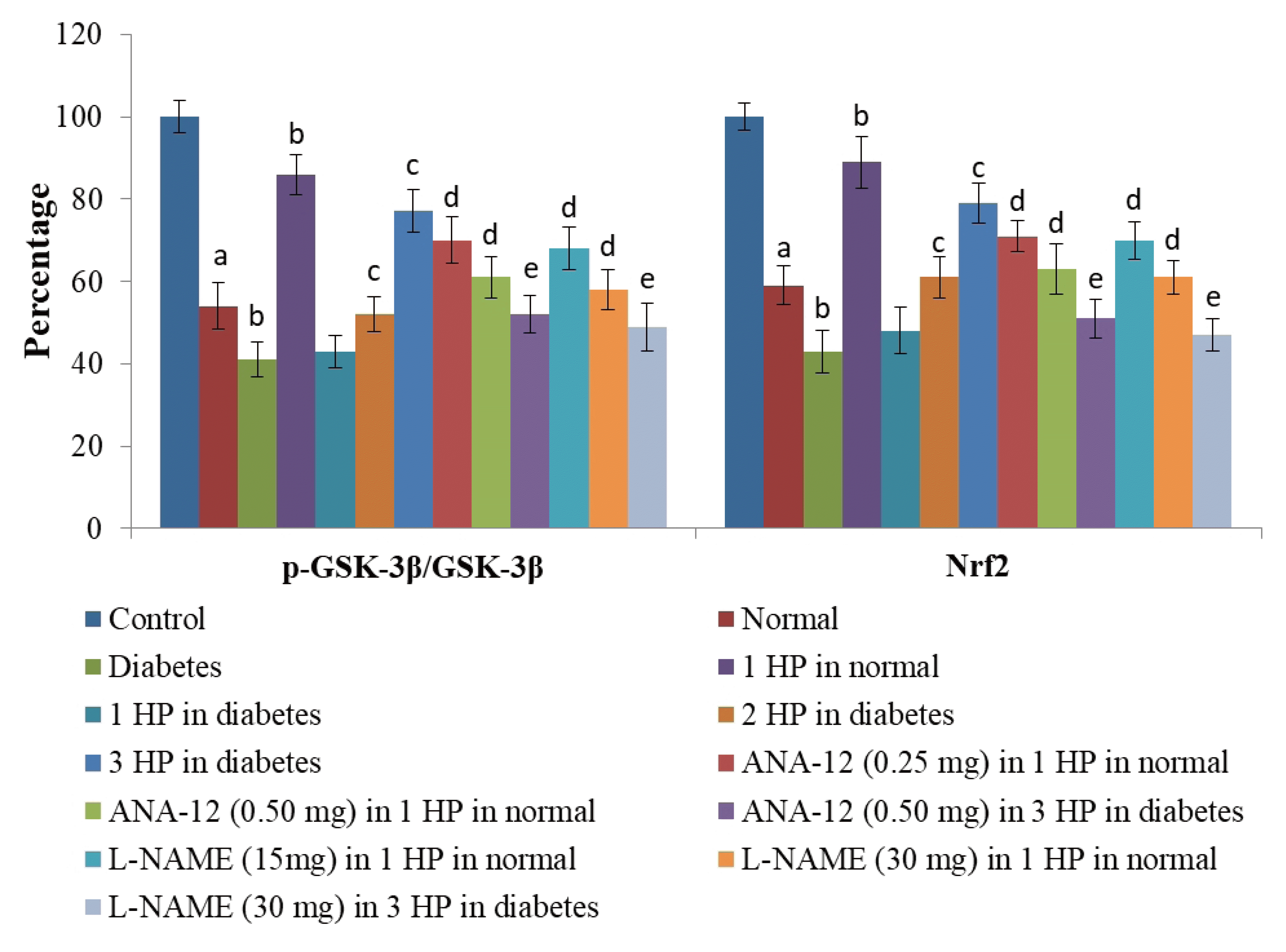 | Fig. 9Effects of different interventions on intracellular signaling pathway i.e., p-GSK-3β/GSK-3β ratio and Nrf2 expression in heart homogenates following ischemia-reperfusion injury.HP, hypoxic preconditioning; 1 HP, single episode of hypoxic preconditioning. ap < 0.05 vs. control; bp < 0.05 vs. normal; cp < 0.05 vs. diabetes; dp < 0.05 vs. 1 HP in normal; ep < 0.05 vs. 3 HP in diabetes.
|
Table 4
Effect of different interventions on brain acetylcholinesterase activity
| S. No | Groups | Brain acetylcholinesterase activity (nM of Ach hydrolysed/min/mg of protein) |
|---|---|---|
| 1 | Normal | 101.2 ± 3.7 |
| 2 | Diabetes | 144.4 ± 3.2a |
| 3 | Single cycle of whole body hypoxic preconditioning in normal | 98.1 ± 3.1 |
| 4 | Single cycle of whole body hypoxic preconditioning in diabetes | 142.1 ± 3.1 |
| 5 | Two cycles of whole body hypoxic preconditioning in diabetes | 132.1 ± 3.8b |
| 6 | Three cycles of whole body hypoxic preconditioning in diabetes | 110.3 ± 3.3b |
| 7 | ANA-12 TrkB antagonist (0.25 mg/kg) in one cycle of whole body hypoxic preconditioning in normal | 99.3 ± 2.3 |
| 8 | ANA-12 TrkB antagonist (0.50 mg/kg) in one cycle of whole body hypoxic preconditioning in normal | 98.9 ± 3.2 |
| 9 | ANA-12 TrkB antagonist (0.50 mg/kg) in three cycles of whole body hypoxic preconditioning in diabetes | 138.4 ± 4.1c |
| 10 | L-NAME (15 mg/kg) in one cycle of whole body hypoxic preconditioning in normal | 97.5 ± 3.2 |
| 11 | L-NAME (30 mg/kg) in one cycle of whole body hypoxic preconditioning in normal | 99.6 ± 3.8 |
| 12 | L-NAME (30 mg/kg) in three cycles of whole body hypoxic preconditioning in diabetes | 139.3 ± 5.1c |
![]()
Pharmacological inhibitors of BDNF and nitric oxide abolishes the beneficial effects of whole-body preconditioning
Administration of ANA-12 (BDNF receptor antagonist) and L-NAME (NO synthase inhibitor) prior to third episode of preconditioning abolished the cognitive restorative functions (increase in ELT and decrease in time spent in target quadrant), improvement in vascular functionality (incomplete relaxation and decrease in pIC50 value) and reduction in cardiac injury (increase in cardiac injury parameters and apoptotic markers) in db/db mice. Administration of ANA-12 and L-NAME prior to preconditioning abolished the reduction in cardiac injury (increase in cardiac injury parameters and apoptotic markers) without significant effect on cognitive and vascular functionality in C57BL/6 mice.
Pharmacological inhibitors of BDNF and nitric oxide modulate biochemical parameters in whole body preconditioning-subjected mice
Administration of ANA-12 (BDNF receptor antagonist) did not modulate the increase in serum nitrite or serum BDNF levels following a single episode of preconditioning in C57BL/6 and three episodes of preconditioning in db/db mice. Administration of L-NAME (NO synthase inhibitor) significantly attenuated the increase in serum nitrite and serum BDNF levels following a single episode of preconditioning in C57BL/6 and three episodes of preconditioning in db/db mice. Administration of ANA-12 and L-NAME significantly abolished preconditioning-induced normalization of p-GSK-3β/GSK-3β ratio and Nrf-2 levels in supernatants of heart homogenates of C57BL/6 and db/db mice following an ischemia-reperfusion injury. Administration of ANA-12 and L-NAME also abolished preconditioning-induced decrease in brain acetylcholinesterase activity in db/db mice without any significant effect in C57BL/6 mice.
Go to :
DISCUSSION
The db/db mouse is a genetic model of type II diabetes mellitus and it exhibits the typical features of human Type 2 diabetes mellitus [33-35]. Therefore, these mice were used to investigate the development of diabetic complications including cognitive impairment, vascular endothelial dysfunction, and enhanced ischemia-reperfusion-induced myocardial injury. C57BL/6 mice of comparable age groups were employed for the comparative study. In the present study, the presence of hyperglycemia in db/db mice confirmed the presence of diabetes mellitus in db/db mice, while normal glucose levels affirmed the non-diabetic state in C57BL/6 mice. In 16th week old db/db mice, there was a significant impairment in cognitive functioning as indicated by no significant decrease in 4th ELT in comparison to 1st day ELT suggesting the impairment in learning (acquiring the information). Moreover, there was a significant decrease in the time spent in target quadrant on 5th day of trial on Morris Water Maze test in db/db mice in comparison to non-diabetic mice suggesting the impairment in memory retrieval function in diabetic mice. The decrease in memory retrieval in these mice is secondary to impairment in learning (acquisition). Biochemically, there was a significant increase in the acetylcholinesterase activity in the brain homogenates of db/db mice in comparison to C57BL/6 mice. Acetylcholinesterase is responsible for the breakdown of acetylcholine, an important neurotransmitter involved in learning and memory. There have been reports showing an increase in acetylcholinesterase activity in diabetic animals [36,37] and impairment in cognitive dysfunction [38,39]. Therefore, it may be possible that an increase in acetylcholinesterase activity during long-standing diabetes may be responsible for the impairment in learning and memory. Moreover, there was impairment in the vascular endothelial functions in db/db mice, assessed on isolated aortic ring segments and an increase in ischemia-reperfusion injury in isolated hearts in comparison to non-diabetic mice. Indeed, there was a significant decrease in pIC50 values along with an increase in cardiac injury parameters (CK-MB, cTnT and LDH-1) and apoptotic parameters (bcl-2 and caspase 3) in db/db mice as compared to C57BL/6 mice. These findings suggesting the impairment in cardiovascular functions during long-standing diabetes mellitus are consistent with earlier published studies [40-42].
Hypoxic preconditioning is the novel intervention in which application of short, intermittent episodes of hypoxia confer tissue protection [43-46]. In whole-body hypoxic preconditioning, the whole animal is exposed to intermittent cycles of hypoxia and this type of preconditioning may be indirectly related to exercise [4]. In the present study, a single episode of whole body preconditioning (on the last day of 15th week) led to a significant reduction in ischemia-reperfusion injury in the isolated hearts of C57BL/6 mice (assessed at the end of 16th week). The application of preconditioning stimuli (ischemic as well as pharmacological) produce immediate as well as delayed, persistent protective effects [47,48] and the protective effects have been shown to last even for several days [49]. In the present study also, the protective effects of whole-body hypoxic preconditioning could be observed even after 1 week and to best of our knowledge, it is the first study documenting the delayed cardioprotective effects (up to 1 week) of whole body preconditioning against ischemia-reperfusion injury. In contrast to the cardioprotective effects of a single episode of whole body hypoxic preconditioning in non-diabetic mice, preconditioning stimulus was not able to confer protection to the hearts of db/db mice against ischemia-reperfusion injury. There have been studies showing that the cardioprotective effects of preconditioning are lost in the diabetic state [50,51]. However, the repeated applications (two or three) of hypoxic preconditioning stimuli in db/db mice exerted the cardioprotection and effects of three episodes of hypoxic preconditioning (at the end of 13th, 14th and 15th week) on myocardial injury were more significant in comparison to two episodes (at the end of 14th and 15th week). Therefore, it is proposed that the loss of beneficial actions of a single episode of preconditioning in diabetic animals may be restored on repeated applications of preconditioning. Similar findings were observed in parameters related to cognitive and vascular endothelial functions. A single episode of hypoxic preconditioning was not capable to prevent vascular dysfunction and cognitive dysfunction in db/db mice. However, three episodes of whole body preconditioning produce led to increase in pIC50 value along with decrease in 4th day ELT and increase in time spent in the target quadrant. Moreover, the repeated cycles of preconditioning also attenuated an increase in acetylcholinesterase activity in the brain homogenates of db/db mice. The studies have shown the beneficial effects of repeated episodes of preconditioning [52]. However, this is the first study documenting that the loss of beneficial effects of whole-body hypoxic preconditioning may be restored in diabetic mice on their repeated applications.
In the present study, the application of a single episode of whole body hypoxic preconditioning in non-diabetic mice increased the serum nitrite and serum BDNF levels, assessed on the same day of application of preconditioning. Since serum BDNF levels are much higher than the plasma BDNF levels [53], therefore, in the present study the levels of BDF were measured in the serum samples. Serum nitrite was used as an indicator of nitric oxide formation. This suggests that the increase in BDNF and nitric oxide may possibly contribute to the beneficial effects of preconditioning in C57BL/6 mice. This contention was supported by the results showing the attenuation of preconditioning-induced cardioprotection by the administration of BDNF antagonist, ANA-12 (TrkB antagonist) and nitric oxide synthase inhibitor, L-NAME prior to application of preconditioning stimulus.
In contrast to non-diabetic mice, the application of a single episode of whole body hypoxic preconditioning failed to increase the serum nitrite and serum BDNF levels in db/db mice. However, repeated episodes of preconditioning led to an increase in serum nitrite and BDNF levels in db/db mice, particularly with three cycles of preconditioning stimuli. It suggests that a single episode may be insufficient to trigger the release of nitric oxide and BDNF and thus, there may be a need for repeated episodes of preconditioning to increase their levels in diabetic animals. Furthermore, BDNF antagonist and nitric oxide synthase inhibitor abolished the cardioprotective effects of three episodes of preconditioning again suggesting the importance of these biochemical mediators in preconditioning-induced cardioprotection. Administration of BDNF antagonist and nitric oxide synthase inhibitor also attenuated the preconditioning-induced decrease in brain acetylcholine activity, improvement in cognition and vascular functionality along within diabetic mice. It further emphasizes the important contribution of BDNF and nitric oxide in repeated episodes of preconditioning-mediated beneficial effects. It is also important to note that the administration of TrkB antagonist (BDNF receptor antagonist) decreased the serum nitrite levels, while L-NAME did not modulate the serum BDNF levels. It possibly suggests that nitric oxide is the upstream mediator of BDNF i.e., an increase in nitric oxide may contribute in increasing the BDNF levels. Amongst the various sources of BDNF, the important role of the endothelium in increasing BDNF production has been emphasized [18,54]. Furthermore, it has been reported that endothelium-derived nitric oxide is important in increasing BDNF levels [18]. Accordingly, it may be suggested that hypoxic preconditioning stimulus may act on the endothelium to increase nitric oxide, which may, in turn, increase the levels of BDNF to produce beneficial effects in non-diabetic as well as in diabetic mice. However, more episodes of preconditioning are needed to increase the BDNF and nitric oxide levels to impart protective effects in diabetic animals.
In the present study, there was also a decrease in the ratio of p-GSK-3β/GSK-3β and Nrf2 in the heart homogenates of db/db as well as C57BL/6 mice following ischemia-reperfusion injury. However, these alterations were more significant in the homogenates of db/db mice. GSK-3β a key part of intracellular signaling cascade and a decrease in phosphorylation indicates its activation [55]. Accordingly, a decrease in the ratio of p-GSK-3β/GSK-3β suggests the increase in the enzymatic activity of GSK-3β. Nrf2 is a transcriptional factor and its decreased levels are indicative of a decrease in protective, antioxidant actions [56]. However, a single episode of hypoxic preconditioning in non-diabetic and three episodes of preconditioning in diabetic mice led to the restoration of p-GSK-3β/GSK-3β ratio and Nrf2 levels suggesting that preconditioning-induced decrease in GSK-3β activity and increase in Nrf2 may contribute in imparting cardioprotection. There have been earlier studies showing the key role of GSK-3β activity [57] and Nrf2 [58] in preconditioning-induced cardioprotection. Administration of BDNF antagonist and nitric oxide synthesis inhibitor attenuated preconditioning-mediated restoration of p-GSK-3β/GSK-3β ratio and Nrf2 levels in C57BL/6 and db/db mice. Accordingly, it may be hypothesized that a single episode (in non-diabetic mice) or three episodes of preconditioning (in diabetic mice) may induce the release of nitric oxide and BDNF that may trigger tissue protection by increasing the p-GSK-3β/GSK-3β ratio and Nrf2 levels.
Along with the use of selective pharmacological blockers, the role of BDNF and nitric oxide in whole body hypoxic preconditioning was affirmed by measuring their levels in the serum following preconditioning stimulus. In future studies, the selective BDNF agonist and nitric oxide donor may be employed to confirm their role in whole body hypoxic preconditioning. Moreover, the employment of selective GSK-3β modulators in the future studies may support the contention of that GSK-3β signaling is important in whole body hypoxic preconditioning-triggered multiorgan protection. Since in this study, a single episode of hypoxic preconditioning conferred protection in normal mice, therefore multiple episodes of hypoxic preconditioning (two and three) were not employed in non-diabetic mice. However, future studies may be designed to explore whether the multiple episodes of hypoxic preconditioning confer additional benefits or alter the biochemical parameters in a significant manner in comparison to a single episode of hypoxic preconditioning.
Repeated episodes of whole body hypoxic preconditioning have the potential to attenuate cognitive impairment, vascular dysfunction and enhancement in ischemia-reperfusion-induced myocardial injury in diabetic mice. It is possible that repeated episodes of hypoxic preconditioning stimuli may increase in the serum levels of nitric oxide and BDNF levels that may eventually activate the GSK-3β and Nrf2 signaling pathway to confer protection.
Go to :
 XML Download
XML Download