

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Hemophagocytic lymphohistocytosis (HLH) is a life–threatening immune disorder associated with widespread lymphocyte and hemophagocytic infiltration of the vital organs and an overwhelming activation of the T-lymphocytes and macrophages [1]. There are two different types of HLH: primary HLH (pHLH) also known as inherited HLH [2] and secondary or acquired HLH (sHLH) [3]. pHLH includes familial HLH (FHL) and primary immunodeficiency-associated HLH. FHL is an autosomal recessive, life-threatening condition characterized by a defective immune response. Molecular evaluations have revealed a diverse pathogenesis for FHL. There are five FHL sub-types (FHL-1 to 5), separated according to genetic mutation caused by germline pathogenic variants in the PRF1, UNC13D, STX11 and STXBP2 genes. In addition, HLH may also be associated with primary immunodeficiency disorders (non-FHL) such as Griscelli syndrome type 2, Chédiak-Higashi syndrome and X-linked lymphoproliferative syndromes (XLP) 1 and 2 caused by germline pathogenic variants in RAB27A, LYST, SH2D1A, and XIAP, respectively. Unfortunately, none of these molecular defects are detected in 10 to 50% of patients and these patients are therefore classified as the unknown-genetics group [2]. Interestingly, ethnic differences in genetic susceptibility to FHL have been reported [1]. Untreated FHL is a fatal disease with a median survival of less than 2 months following diagnosis [4]. The establishment of diagnostic criteria and the use of immunochemotherapy with or without appropriate allogeneic hematopoietic stem cell transplantation (HSCT) have significantly improved survival outcomes in patients with HLH [5]. The spectrum and frequency of germline pathogenic variants in the HLH genes of the Saudi population is unknown. Therefore, this study was designed to explore the spectrum of inherited variants of the eight FHL related genes in a large cohort of Saudi FHL patients.
MATERIALS AND METHODS
Patients
A total of 87 pediatric Saudi patients diagnosed with HLH between January 1995 and December 2014 at the King Faisal Specialist Hospital and Research Centre (KFSHRC) in Riyadh, Saudi Arabia, were enrolled in this study. All patients met the HLH-2004 diagnostic criteria as described by the Histiocyte Society [6]. Clinical data including demographic characteristics, family history, and treatment outcomes were obtained through the review of their medical records and the patients were treated according to either the HLH-1994 or HLH-2004 protocol. A written informed consent to obtain blood samples or to use stored DNA samples was obtained from each patient or their parent. A waiver for this consent was granted by the Research Advisory Committee of our institution in the case of samples from deceased patients. This study was approved by our hospital’s IRB and funded by King Abdulaziz City for Science and Technology, Saudi Arabia.
DNA isolation
Blood samples from parents, patients and/or their siblings were collected when available at the time of diagnosis. For expired patients or for those who had undergone HSCT by the time of this study, DNA specimens were collected from the HLA typing lab. DNA was isolated from peripheral blood leukocytes using standard genomic DNA purification methods.
PCR and direct sequencing analysis
PCR and direct sequencing were used to identify the germline pathogenic variants in both the exons and splicing regions of PRF1, UNC13D, STX11, STXBP2, LYST, RAB27A, SH2D1A and XIAP, as well as the deep intronic regions of UNC13D covering the c.118-308 and c.118-307 loci. These evaluations were completed as described in the Supplement Materials and Methods (Supplement 1). The sequencing raw data were analyzed using DNA Seqman (Lasergene 6) and all the reference sequences were downloaded from GenBank.
Assessment of pathogenicity of variants
ACMG/AMP guidelines and literature evidence were used to support the interpretation of the impact of the sequence variants identified in this study [7]. All novel variants of uncertain significance (VUSs) as identified by ACMG/AMP were further analyzed using four in-silico pathogenicity prediction tools: PolyPhen-2, SIFT, CADD and MutationTaster. The variants predicted as damaging or possibly damaging by at least three of these in-silico prediction tools were assigned as likely to be pathogenic variants.
Definitions
We used the HLH diagnostic criteria established by the Histiocyte Society [6]. Central Nervous System (CNS) involvement was defined as the development of neurological signs and/or symptoms, abnormal cerebrospinal fluid (pleocytosis and/or elevated protein), and neuroradiological findings [8]. In our study, patients harboring pathogenic variants in PRF1, UNC13D, STX11 and STXBP2 were designated as FHL, while patients carrying pathogenic variants in LYST, RAB27A, SH2D1A and XIAP were designated as non-FHL and patients in whom genetic screening did not reveal any genetic defects were designated as the unknown-genetics group. Overall survival (OS) was defined as survival from the date of diagnosis to last follow-up or death and death from any cause was recorded as a single event. The last follow-up was carried
Statistical evaluations
IBM-SPSS version 20.0 (IBM, Armonk, USA) was used for all statistical analyses. Pearson’s chi-square test was used to determine whether there was a difference in qualitative variables between groups and OS was estimated using Kaplan-Meier survival analysis and compared using an appropriate test. The results were considered statistically significant at P<0.05.
RESULTS
Of the 87 patients analyzed, 62 (71.3%) were categorized as FHL, 7 (8.0%) as non-FHL group, and 18 (20.7%) as part of the unknown-genetics group. The median age at presentation was 0.45 years (range, 0–8.5 yr). Patients in the FHL group presented at a significantly younger age than those with unknown-genetics or non-FHL disease (median, 0.3 vs. 0.8 vs. 3.2 yr, respectively; P=0.005). A total of 28 (32%) patients presented in the first 3 months of life while 63 (72%) were under the age of one at the time of diagnosis. Only 9.6% (6 out of 62) of the FHL patients experienced disease onset beyond 2 years of life. Notably, the majority of the infant patients (50 of 63, 79%) were shown to have FHL (Table 1). The demographic data and clinical features for all of the patients diagnosed with HLH at their initial presentation are provided in Table 2. A history of parental consanguinity was present in 67 (77%) patients while there was a history of siblings with HLH in 22 (25.2%) patients. CNS involvement at diagnosis was detected in 67.3% of the 52 evaluable patients and the cumulative probability of OS at 5-years was 55.4% with a median follow-up of 5.6 years (range, 0.1–20.9 yr). There were no significant differences in OS across age groups at presentation (≤12 mo vs. more), CNS disease (presence vs. absence) or gender. However, FHL group (N=62), patients with PRF1 mutations had a significantly poorer 5-year OS (21.4%) when compared to STX11 (47.4%), STXBP2 (57.4%) and UNC13D (80.8%, P=0.008, Table 2). A total of 24 (27.6%) children died following disease progression prior to HSCT at a median duration of 0.8 years (0.05–5.35 yr), leaving 63 (72.4%) patients who underwent HSCT. Patients who received HSCT had a significantly better 5-year OS than those without HSCT (66.5±6.2% vs. 0%, P<0.001, Table 2, Fig. 1). No statistically significant difference was found for 5-year OS among the FHL, non-FHL or unknown-genetics groups for those patients who underwent HSCT (P=0.984, Table 2, Fig. 2).
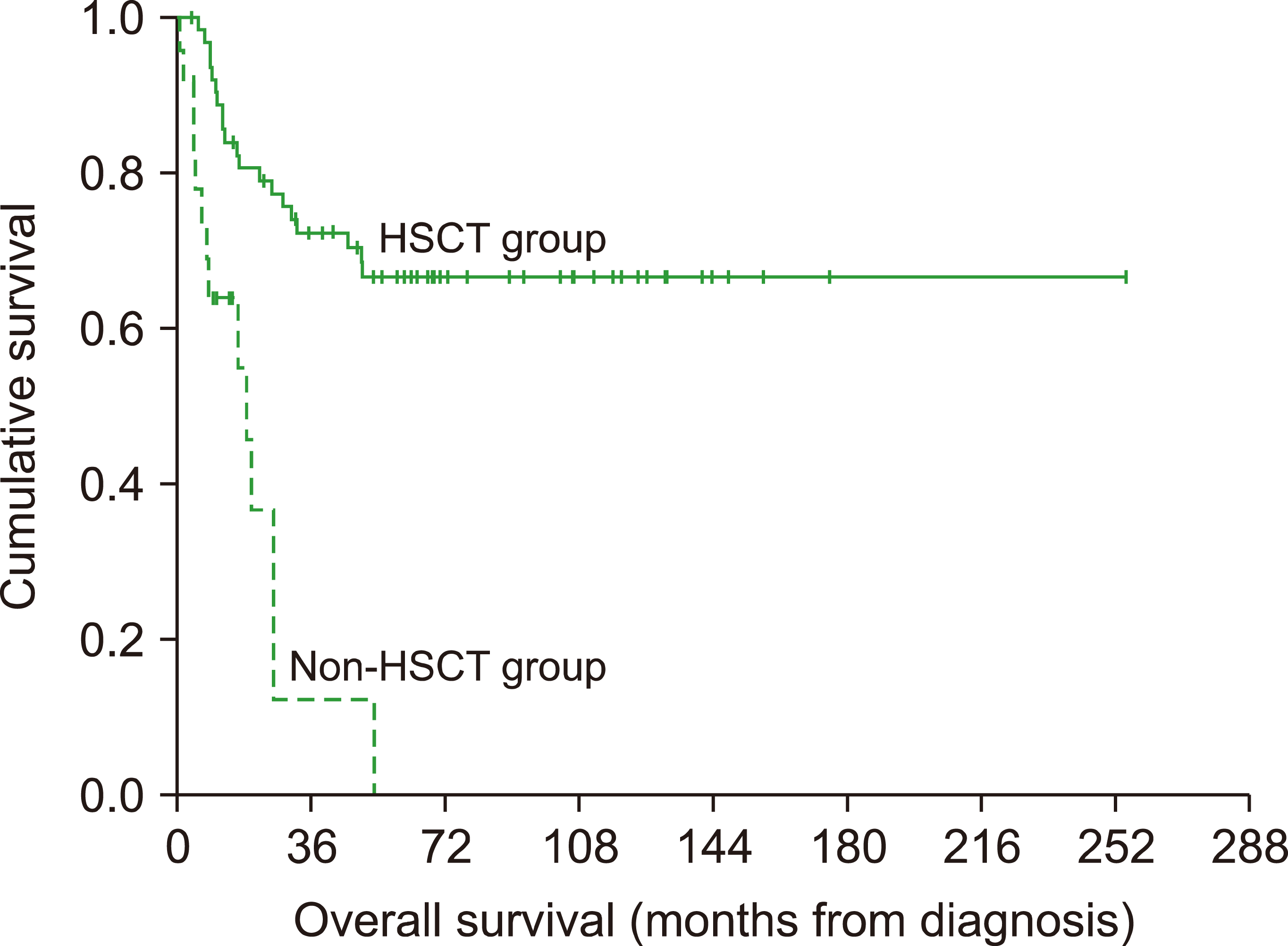
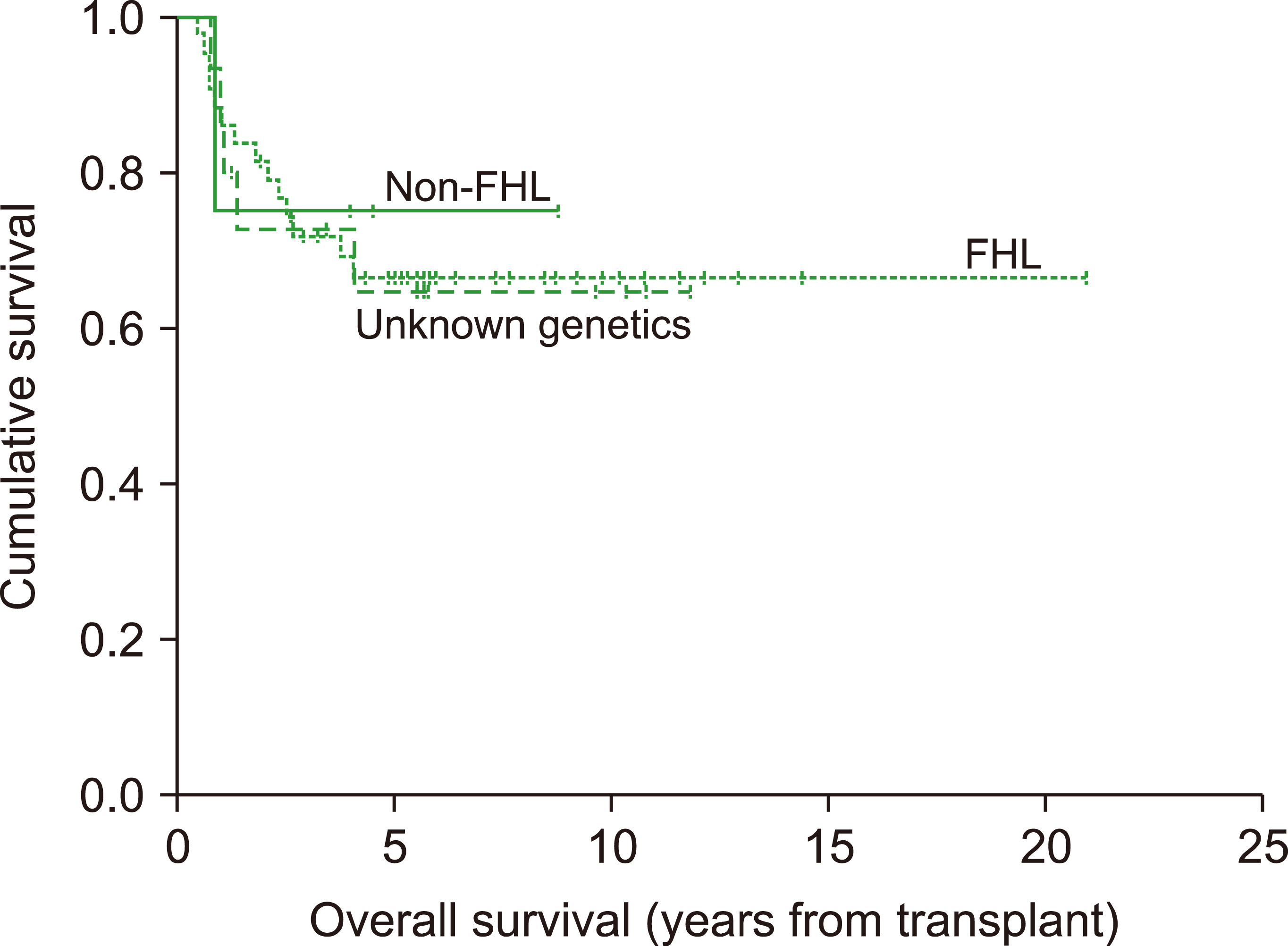
Table 1
Frequency of FHL-associated gene mutations by age (N=87).
![]()
Table 2
Clinical and laboratory characteristics, CNS involvement at presentation and clinical outcome for patients in different HLH groups.
![]()
Mutations in HLH-related genes
The analysis of the 87 patients in this cohort revealed a total of 20 pathogenic variants in seven different genes in 69 patients (79.3%), including eight previously reported variants and 12 novel variants (Table 3, Supplementary Figs. 1–5). Among the 12 novel variants, five were frame shift mutations, resulting in stop/gain or splicing site variants causing a loss of protein function and identified as likely to be pathogenic according to the ACMG/AMP guideline. The remaining seven missense variants were interpreted as having uncertain significance using the ACMG guideline due to the lack of functional evaluation. Given this we went on to evaluate these variants using four computational prediction tools. Among the seven missense variants, six were identified as deleterious by all four prediction tools while only one variant (c.1034 C>T, p. Thr345Met) was identified as deleterious in only three tools. In addition, our clinical data showed that all of these missense variants were unique to the patients and none were detected in unaffected family members. Moreover, these seven missenses variants were absent in the 250 healthy controls used in this study. In addition, 77% patients presented with parental consanguinity and 53% of these patients had affected siblings. Taken together these data strongly imply that these seven missense variants are likely to be pathogenic. However, functional studies are required to confirm the pathogenicity of these variants (Supplementary Table 1).
Table 3
HLH-associated genetic mutations (N=69).
| Gene | Nucleotide change | Amino acid change | Mutation type | Novelty | N of Patients | Age at onset (mo), median, range | Patient status (alive/dead) | Reference |
|---|---|---|---|---|---|---|---|---|
| PRF1 | c.673C>T | p.(Arg225Trp) | Missense | Reported | 1 | 83.9 | 0/1 | Stepp et al. [33] |
| c.1122G>A | p.(Trp374*) | Nonsense | Reported | 3 | 2.6 (0–6.0) | 1/2 | Balta et al. [24] | |
| c.50delT | p.(Leu17ArgfsTer34) | Frameshift | Reported | 2 | 2.7 (2.3–3.0) | 0/2 | Trizzino et al. [34] | |
| c.1081A>T | p.(Arg361Trp) | Missense | Reported | 1 | 11.4 | 0/1 | Gadoury-Levesque et al. [35] | |
| UNC13D | c.1484T>G | p.(Val495Gly) | Missense | Novel | 1 | 72.4 | 1/0 | This study, Supplementary Fig. 1C |
| c.3047_3048insC | p.(Glu1017ArgfsTer8) | Frameshift | Novel | 6 | 2.4 (1.9–11.6) | 4/2 | This study, Supplementary Fig. 1A | |
| c.3053C>A | p.(Ala1018Asp) | Missense | Novel | 2 | 2.6 (1.9–3.3 ) | 2/0 | This study, Supplementary Fig. 1D | |
| c.2019C>G | p.(Tyr673*) | Nonsense | Novel | 1 | 15.1 | 1/0 | This study, Supplementary Fig. 1B | |
| c.766C>T | p.(Arg256*) | Nonsense | Reported | 2 | 1.9 (1.6–2.1) | 2/0 | Zur Stadt et al. [19] | |
| STXBP2 | c.862T>C | p.(Trp288Arg) | Missense | Novel | 1 | 0.43 | 0/1 | This study, Supplementary Fig. 2 |
| c.1430C>T | p.(Pro477Leu) | Missense | Reported | 24 | 3.7 (0.8–14.2) | 16/8 | zur Stadt et al. [15] | |
| c.1034C>T | p.(Thr345Met) (monoallelic) | Missense | Novel | 2 | 7.2 (5.3–9.0) | 0/2 | This study | |
| STX11 | c.601_602ins C | p.(Gln140ProfsTer46) | Frameshift | Novel | 1 | 16.9 | 1/0 | This study, Supplementary Fig. 3 |
| c.690G>A | p.(Gln230Ala) (monoallelic) | Missense | Novel | 1 | 9.5 | 0/1 | This study | |
| c.173T>C | p.(Leu58Pro) | Missense | Reported | 14 | 12.1 (0.7–63.6) | 9/5 | Zur Stadt et al. [19] | |
| LYST | c.4637C>T | p.(Ala1546Val) | Missense | Novel | 1 | 10.9 | 0/1 | This study, Supplementary Fig. 4C |
| c.9560+1G>C | Splice site | Splice site | Novel | 1 | 46.9 | 1/0 | This study, Supplementary Fig. 4B | |
| c.9044+1G>T | Splice site | Splice site | Novel | 2 | 59.9 (47.0–72.9) | 2/0 | This study, Supplementary Fig. 4A | |
| RAB27A | c.400A>C | p.(Lys134Gln) | Missense | Novel | 2 | 12.0 (8.0–16.0) | 1/1 | This study, Supplementary Fig. 5 |
| XIAP | c.1190insA | p. (Ile397Asnfs Ter405) | Frameshift | Reported | 1 | 39.3 | 0/1 | Aguilar et al. [36] |
![]()
Of the 69 patients harboring pathogenic variants, 65 (94.2%) carried homozygous variants, 25 (36%) in STXBP2, 15 (21.7%) in STX11, 12 (17.5%) in UNC13D, 7 (10%) in PRF1, 4 (5.8%) in LYST and 2 (3%) in RAB27A respectively. Strikingly, almost one third of patients (30.4%, 21 of 69) harbored novel pathogenic variants including 10 in UNC13D, 4 in LYST, 3 in STXBP2, 2 in STX11 and 2 in RAB27A accounting for 14.5%, 5.8%, 4.3%, 2.9% and 2.9% of all mutant patients respectively. This data highlights the specific spectrum of pathogenic variants in our population.
Patients with FHL
STXBP2 mutations: We isolated three different STXBP2 missense mutations (Table 3) in the 27 FHL-5 patients identified in this study, and 25 of these patients were homozygous for their STXBP2 mutation. The most frequent mutation was (c.1430C>T, p. Pro477Leu), which was detected in 24 (89%) patients. A homozygous (c.862 T>C, p. Trp288Arg), and a mono-allelic (c.1034 C>T, p. Thr345Met) novel missense mutation were also observed in one and two patients, respectively. The median age at diagnosis was 0.3 years (range, 0.08–1.17) and was shown to be 0.3 years for homozygotes and 0.6 years for heterozygotes. Notably, 21 (78%) of the STXBP2 patients originated from the same geographical area and 16 (59%) of these patients belonged to the same tribe.
STX11 mutations: We found 3 different STX11 mutations in the 16 FHL-4 patients within our cohort (Table 3). The most frequent mutation was shown to be missense, c.173T>C (p. Leu58Pro), which was present in 14 (87.5%) patients. We also detected a novel mutation; c.601_602ins C (p. Gln140ProfsTer46) in one patient and another heterozygous mutation, c.690 G>A (p. Gln230Ala), in the remaining patient who died from disease progression. The median age at presentation was 1.0 year (range, 0.05–5.2) and was 0.78 years in the patient with a mono-allelic mutation. Like in STXBP2 patients, consanguinity was very high (75%) and all patients originated from a single tribe from a discrete geographical area.
UNC13D mutations: We found 5 different UNC13D mutations in the FHL-3 patients (Table 3). There were four novel mutations in this group of ten patients, two of which resulted in the insertion of a stop codon. The most prevalent mutation was c.3048_3049insC (p.Glu1017ArgfsTer8), which was found in six patients, two of whom were siblings while the other four patients were from different families. The second novel mutation was missense mutation, c.3053C>A (p. Ala1018Asp)), which was found in a brother and his sister while the two remaining novel mutations; c.1484T>G (p. Val495Gly) and c.2019C>G (p. Tyr673*), were found in two unrelated boys. A previously reported mutation: c.766 C>T (p. Arg256*), was found in the remaining two patients. The median age at diagnosis was 0.2 years (range, 0.13–5.95).
PRF1 mutations: The most frequent PRF1 mutations were c.1122G>A (p. Trp374*) which was present in three patients and c.50delT (p.Leu17ArgfsTer34) which was found in two patients. The median age at presentation was 0.24 years (range, 0–6.9) and unlike in the FHL-4 and FHL-5 patients, no specific geographical pattern was observed in either the FHL-2 or FHL-3 patients.
Patients with non-FHL HHL
In the 7 patients presenting with mutations in non-classical FHL genes, two novel mutations were detected in the LYST gene in four patients confirming their diagnosis with Chédiak -Higashi syndrome. The most common mutation was c.9044+1G>T (splice site) which was found in three samples, with two of these being siblings. Two siblings were diagnosed as Griscelli syndrome after detecting a missense mutation, c.400A>C (p. Lys134Gln) in the RAB27A gene. The remaining patient was found to have a c.1190insA (p.Ile397 AsnfsTer405) mutation in XIAP. Unlike the FHL patients, the median age at diagnosis was higher in the Non-FHL group; 3.2 years (range, 0.67–5.99) (Table 2) and the index cases of Griscelli syndrome type 2 or Chédiak-Higashi syndrome were initially diagnosed as having FHL due to a lack of the classical clinical features in these patients.
Patients with unknown-genetics
These patients were assumed to have HLH based on the presence of consanguinity, familial recurrence or disease reactivation. The 18 patients with unknown-genetic etiology presented with a median age at disease onset of 0.7 years (range, 0–8.53) with a median survival of 3.8 years (range, 0.7–11.8). A history of affected family members and consanguinity was observed in 7 (40%) and 11 of the 18 (68.7%) patients, respectively. CNS involvement and disease reactivation were observed in 6 of 12 (50%) and 5 (27.8%) patients, respectively (Table 2).
DISCUSSION
Identification of population specific genetic markers for HLH is very important as it allows early recognition of HLH and facilitates earlier HSCT as well as the screening of family donors for HLH. Although FHL is a very rare disorder, it is relatively common in the Saudi population, likely because of the frequent consanguineous marriages in this population. The spectrum of HLH gene mutations in this population is unknown. This is, to our knowledge, the first report to comprehensively describe the mutations present in FHL-associated genes and their clinical characteristics in the Saudi population. FHL is usually diagnosed during infancy or early childhood. Our analysis revealed a median age at diagnosis of FHL of 0.4 years, with 79% of the patients being infants. In fact, only 4.8% of our FHL patients presented with disease onset at over 5 years of age which is comparable to the incidence rates recorded by the International Histiocyte Society which reports that approximately 5% of patients are affected after 5 years of age [9]. Previous studies have reported that adolescents and even older adults may have a genetic predisposition to HLH [10-13]. Our findings confirmed that STXBP2 is the most commonly mutated gene (36%) in Saudi patients with FHL. This is in contrast with Elyamany et al. whose single-center study of 12 Saudi patients reported mutations in the STX11 gene (5 of 10, 50%) and PRF1 gene (2 of 10, 20%) were reported and only one patient with an abnormality in STXBP2 [14]. Our institution is the largest tertiary care facility in the country. Therefore, we believe our patient cohort represent a more global population. The most frequent mutation in the FHL-5 patients was (c.1430C>T, p. Pro477Leu) which was observed in all but one (96%) homozygous FHL-5 patient. To date, the c.1430C>T, p. Pro477Leu mutation has only been observed in patients of Saudi or Kuwaiti origin [15, 16]. Most of the reported STXBP2 patients, including ours, originate from Saudi Arabia (Table 4). In FHL-5 families with more than one affected child, we noted that intestinal symptoms were either present in all siblings or absent in all affected siblings, suggesting that the presence or absence of intestinal symptoms in FHL-5 patients is related to family-specific STXBP2 mutations. Chronic diarrhea has been reported in STXBP2 patients, in addition to other atypical features, including sensorineural hearing loss and abnormal bleeding [17]. Interestingly, none of these atypical features were detected in any of our STXBP2 patients. However, Baothman et al. [18] recently published a report on a Saudi girl with HLH who experienced chronic diarrhea since birth. All three of her deceased siblings had a similar clinical presentation. Whole-exome sequencing analysis of this patient revealed a homozygous missense mutation, c.1139A>G, p.G380A, in STXBP2.
Table 4
![]()
STX11 is the second most commonly mutated gene (21.7%) in Saudi FHL patients. Interestingly, 14 of 16 patients with STX11 mutations originated from a single tribe harboring a c.173T>C (p. Leu58Pro) mutation. Although, STX11 mutations were previously thought to be restricted to patients originating from Turkey [19], our study shows that our FHL-4 cohort is more common in our population than in the previously reported Turkish ethnic group (Table 4). Interestingly, both STXBP2 and STX11 patients exhibited a distinct geographical association. This can be explained by the high prevalence of consanguinity which was previously reported by El‐Mouzan et al. [20] with consanguinity observed in as much 56% of the Saudi population compared to the roughly 10% consanguinity of the global population. Although Oman is a neighboring country in the Arabian Peninsula, the most commonly mutated FHL gene in this region is PRF1 [21] possibly due to ethnic differences in the genetic predisposition of each population to each subtype of FHL. FHL-5 and FHL-4 are the most common FHL subtypes in our cohort and account for 58% of the FHL cases in our study as opposed to studies in southern Europe where FHL-2 and FHL-3 account for 70% of cases. In addition, the most commonly mutated gene in Japanese and Chinese FHL patients in PRF1 [22]; whereas FHL-3 is the predominant causative gene in the Korean population [23].
Of the five different UNC13D mutations observed in ten unrelated families, four were not reported previously. The most prevalent mutation was c.3048_3049insC (p. Glu1017ArgfsTer8) which was found in 50% of the patients. These patients experienced early onset of disease (median 2.4 mo of age) and only two survived after HSCT. Although the c.766 C>T (p. Arg256*) nonsense mutation was previously reported as a heterozygous mutation [19], it was found as a homozygous mutation in two patients from our cohort. FHL-2 is commonly associated with a more severe clinical phenotype which leads to death in a very short period of time, often even before starting specific HLH protocol therapies [24]. This agrees with our FHL-2 cohort data where all but one of the patients in this group expired prior to HSCT. Although, the c.1122G>A (p. Trp374*) and c.50delT (p. Leu17ArgfsTer34) mutations in the PRF1 gene were reported to be primarily associated with Turkish populations [19, 24] and African-Americans, respectively, we observed these mutations in three and two patients in our cohort, respectively.
There is growing evidence that monoallelic variants can also contribute to FHL. In the Cetica et al. study [25], 43 (18%) of their 240 patients presenting with sporadic HLH had monoallelic mutations in one of the FHL-related genes. WES of patients with rheumatoid disorders experiencing MAS, where found to carry several variants in FHL-related genes, as well as other novel candidate genes [26]. Interestingly, our three monoallelic FHL patients (Table 3) presented with a severe disease phenotype as evidenced by the early disease presentation (median, 0.7 yr) and death due to disease progression. The fact that some patients with FHL have been found to have a monoallelic variant in one of HLH-associated genes suggests that there may be other types of pathogenic variants in other alleles which are not detected using traditional sequencing approaches or that these patients harbor pathogenic variants in additional genes that contribute to the development of HLH [27]. This is supported by the fact that between 10 and 50% of HLH patients present with no obvious molecular defects [2, 28]. One major finding of our study is that genetic mutations were not detected by direct sequencing in 20% of patients; who are now being evaluated by whole-exome sequencing. The most likely explanation is that these patients might have mutations in 1 or more gene(s) that have not yet been identified as FHL related, as confirmed by Chinn et al. [29] who demonstrated that only 19 (19%) of their 101 subjects presented with biallelic FHL gene defects. Furthermore, in 28 (58%) of 48 subjects, whole-exome sequencing analyses successfully identified likely molecular explanations, including underlying primary immunodeficiency diseases, dysregulated immune activation and proliferation disorders, and potentially novel genetic conditions. In our non-FHL cohort, the index cases of Griscelli syndrome type 2 or Chédiak-Higashi syndrome were initially diagnosed as having FHL due to the lack of classical clinical features in these patients. Thus, the clinical diagnosis of some patients with Griscelli or Chédiak–Higashi syndromes may be overlooked, particularly in fair-haired patients with HLH [30]. According to the literature, 70% of FHL patients are diagnosed in the first year of life. Our analysis revealed a median age at presentation of 5.3 months with 72.4% of the patients diagnosed before the age of one. The proportion of infants younger than 12 months was significantly higher in the FHL group (P=0.005) and when we analyzed the individual FHL subgroups, we found that the median age of disease onset was higher (12.1 mo) in patients with STX11 mutations than in the other subgroups; PRF1 (3 mo), STXBP2 (3.7 mo) and UNC13D (2.4 mo; P=0.028, Table 2). This concurs with Rudd et al. [31] who reported STX11 mutations in 6 out of 34 patients, half of whom were diagnosed after one year of age.
CNS involvement at the time of diagnosis was relatively common in our patient cohort, which agrees with most of the available literature. Although it is a well-known fact that CNS involvement has a negative effect on FHL prognosis [8], we did not find any differences between the survival rates of patients with and without CNS involvement in different FHL groups. Overall, 24 (27.6%) of our patients died from disease progression prior to HSCT at a median duration of 9.7 months with the majority of these fatalities presenting with PRF1 mutations. This is similar to the HLH-1994 study [5] in which 25 (22%) patients succumbed to disease progression prior to HSCT. Our study confirms that HSCT is the only curative option for FHL patients as we found that patients undergoing HSCT fared significantly better in terms of their 5-year OS than their counterparts (66.5±6.2% vs. 0%, P<0.001, Table 2). For those patients who underwent HSCT, no statistically significant difference was found for the 5-year OS of FHL, non-FHL or unknown-genetics patients (Table 2).
In conclusion, our study demonstrates the unique FHL profile in the Saudi population and highlights the tribal and geographical patterns of STXBP2 and STX11 mutations in Saudi patients with FHL. Thus, the variant profile of Saudi FHL patients is quite different from that of Western and Asian cohorts in which, PRF1 (FHL-2) and UNC13D (FHL-3) are predominate. Our data confirms that HSCT is a life-saving modality for the treatment of FHL patients.
 XML Download
XML Download