

 PDF
PDF Citation
Citation Print
Print



Dear Editor,
The advent of next-generation sequencing (NGS) techniques has considerably improved the detection and characterization of microbial and viral genetic sequences. Although initially used to study environmental, animal, and human samples, NGS has been more recently introduced in clinical microbiology and virology laboratories [1–5]. Its technical requirements, processing time, and cost are now compatible with diagnostic use. NGS allows identification of infectious agents based on complete genomes and not only genes or gene fragments. Particularly the Oxford Nanopore Technology (ONT; Oxford, UK) permits extremely promising sequencing speed [3, 6]. ONT allows single-molecule sequencing of DNA or RNA fragments. Native DNA or RNA molecules are pulled through nanoscale pores one molecule at a time. While the nucleic acid passes through the pore, sensors detect changes in the ionic current that are specific to each passing nucleotide, which allows to determine the nucleic acid sequence.
While NGS is generally used following specific target enrichment, it can be applied directly to nucleic acids extracted from clinical samples. We recently used the latter approach in our clinical microbiology/virology laboratory (IHU Méditerranée Infection, Marseille) to detect and characterize genomes of hepatitis B virus (HBV) by Illumina NGS (Illumina, Inc., San Diego, CA, USA) for the first time and of SARS-CoV-2 by both Illumina and ONT NGS [4, 7]. Compared with Sanger population sequencing, NGS provides information on mutations, deletions, and insertions for all viral genes simultaneously as well as for intergenic regions, and therefore allows a more comprehensive and deeper assessment of viral genomes. Such comprehensive data can contribute to the elucidation of the genome features associated with various clinical characteristics, which, in the case of HBV infection, include acute liver failure, drug resistance, immune escape, or viral reactivation. Here, we describe NGS of a full-length HBV genome and a partial hepatitis delta virus (HDV) genome directly from a single patient plasma sample using ONT GridION. The ethics committee of IHU Méditerranée Infection, Marseille, France, approved this study (No. 2019-001).
In June 2020, we obtained a plasma sample from a 27-year-old male patient to monitor his HBV and HDV infections diagnosed in October 2018 during a systematic screening. He was not aware of viral hepatitis in his family. It is unknown whether he was co-infected with HBV and HDV or experienced HDV superinfection on chronic HBV infection. In November 2018, liver ultrasound testing showed normal results, transient elastography showed no liver fibrosis, and the patient was clinically asymptomatic.
In June 2020, ALT and AST levels were 87 IU/L and 115 IU/L, respectively (normal upper values, 50 IU/L). Serological testing showed hepatitis B surface (HBs) antigen (Ag)-positivity, total anti-HB core antibody (Ab)-positivity, anti-HBs Ab-negativity, hepatitis B e (HBe)Ag-positivity, and anti-HBe Ab-negativity (Architect assays, Abbott Diagnostics, Mannheim, Germany). The serum HBV DNA load was 8.0 log10 IU/mL (NeuMoDx HBV Quant Assay, Qiagen, Courtaboeuf, France). Anti-HDV IgG and IgM testing (Euroimmun, Lubeck, Germany) were both positive, and the HDV RNA load determined by an in-house PCR assay as previously described [8] was 9.6 log10 copies/mL. We used ONT GridION as previously described [7] to obtain HBV and HDV genome sequences without pre-PCR amplification. Briefly, DNA and RNA were extracted from 200 μL of plasma using the EZ1 Virus Mini Kit v2.0 on a BioRobot EZ1 workstation (Qiagen), and RNA was reverse-transcribed using SuperScript IV (Thermo Fisher Scientific, Waltham, MA, USA) before cDNA second-strand synthesis with Klenow Fragment DNA polymerase (New England Biolabs, Beverly, MA, USA). The DNA was then purified using Agencourt AMPure XP beads (Beckman Coulter, Villepinte, France) and sequenced using the ONT Ligation Sequencing Kit on a GridION X5 instrument (Oxford Nanopore Technologies Ltd.). Genome consensus sequences were generated with the CLC Genomics Workbench v.7 (https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-clc-genomics-workbench/) by mapping to HBV and HDV reference genomes (GenBank accession numbers M32138 and M84917, respectively), with 0.85% similarity and 50% coverage as thresholds. All data were generated as part of the routine study at Assistance Publique-Hôpitaux de Marseille (APHM), and this study results from routine standard clinical management. Access to the patients’ biological and registry data issued from the hospital information system was approved by the data protection committee of APHM and was recorded in the European General Data Protection Regulation registry under number RGPD/APHM 2019-73.
In total, 41 (average length, 918 nucleotides) and three (average length, 490 nucleotides) reads were identified as belonging to HBV and HDV, respectively, generating full-length HBV and partial HDV genomes with a size of 3,179 and 326 nucleotides, respectively (available from https://www.mediterranee-infection.com/acces-ressources/donnees-pour-articles/hbv-hdv-ngs/). Phylogenetic analyses performed using the MEGA6 software (http://www.megasoftware.net/) identified HBV of genotype D and HDV of genotype 1 (Fig. 1). The mean diversity per nucleotide position of the HBV genome was 5.4%±9.0%, and the mean coverage was 8.9±2.9. Neither anti-HBV drug resistance mutations nor HBsAg escape variants were detected. A 570-nucleotide-long sequence corresponding to the HDV Ag-encoding gene generated by Sanger population sequencing as previously described [8] clustered with that obtained using ONT GridION in the phylogenetic tree (Fig. 1).
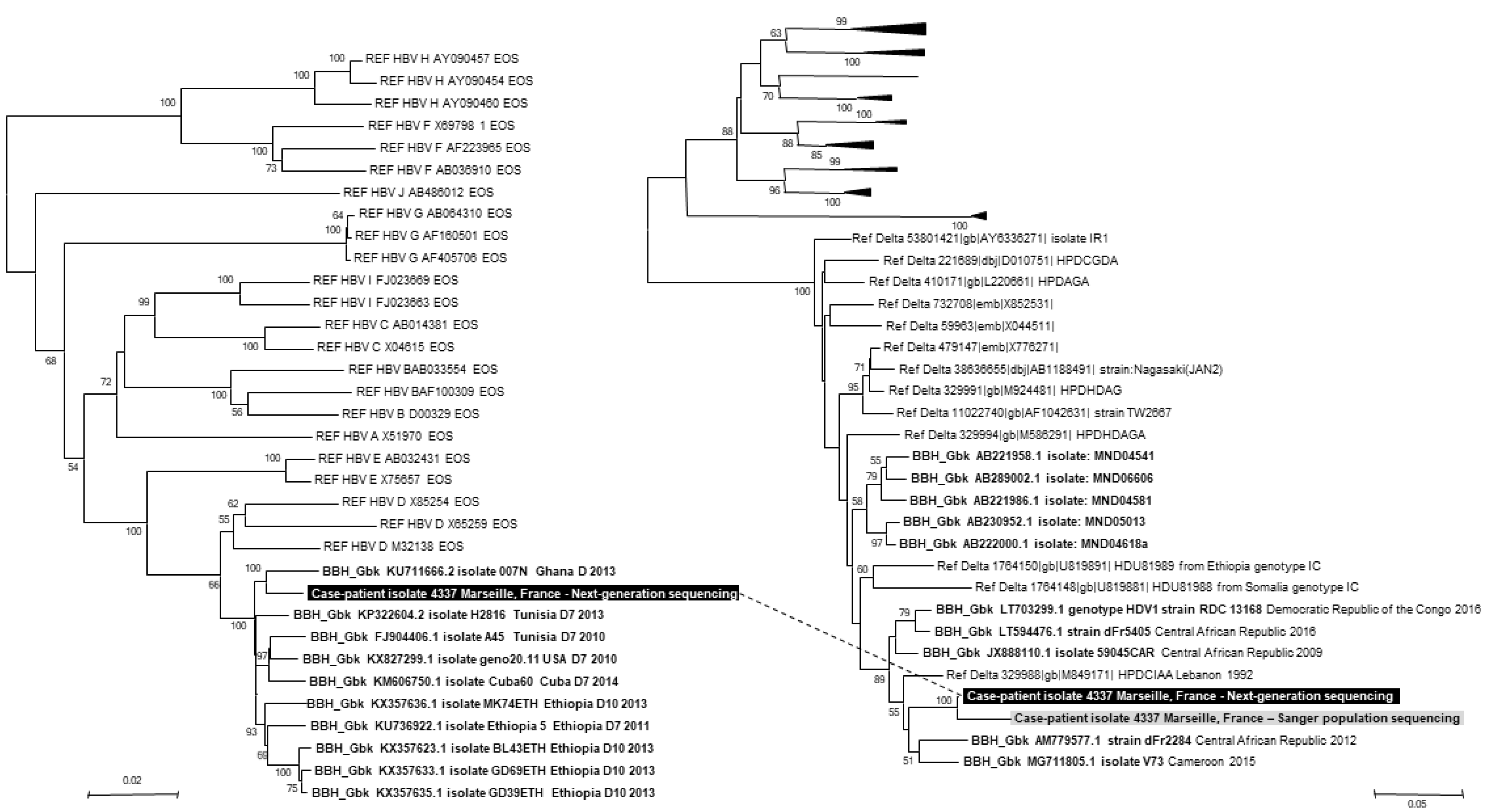 | Fig. 1Phylogenetic trees based on the HBV genome and HDV antigen-encoding gene. Phylogenetic reconstruction based on HBV genome sequences involved 3,252 positions in the final dataset, and phylogenetic reconstruction based on the HDV antigen-encoding gene involved 343 positions corresponding to nucleotides 824–1,154 of the HDV genome (GenBank accession no. AM779577.1). Viral sequences obtained from the patient’s plasma using ONT (available from https://www.mediterranee-infection.com/acces-ressources/donnees-pour-articles/) are indicated by a white bold font on a black background, and those obtained by Sanger population sequencing are indicated by a black bold font on a grey background. HBV and HDV sequences obtained from the patient using ONT are connected by a dashed line. The 10 sequences with the highest BLAST scores recovered from the NCBI GenBank nucleotide sequence database (http://www.ncbi.nlm.nih.gov/nucleotide/), labeled with BBH GbK and indicated by a black bold font, were incorporated in the phylogeny reconstructions, in addition to reference sequences (indicated by Ref for Reference) for HBV or HDV genotypes. Nucleotide alignments were performed using the MUSCLE software (http://www.ebi.ac.uk/Tools/msa/muscle/). Evolutionary history was inferred using the MEGA6 software (http://www.megasoftware.net/) using the neighbor-joining method and the Kimura 2-parameter method. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1,000 replicates) is shown next to the branches. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree; the scale bars indicate the number of nucleotide substitutions per site. Bootstrap values >50% are indicated on the tree.
Abbreviations: HBV, hepatitis B virus; ONT, Oxford Nanopore Technology; NCBI, National Center for Biotechnology Information; BBH GbK, best BLAST hit GenBank.
|
We report for the first time, to the best of our knowledge, the concurrent detection and determination of HBV and HDV sequences, including full HBV genome recovery, on the same day and directly from nucleic acids extracted from plasma. Illumina or ONT NGS has been previously used to obtain HBV or HDV sequences, but generally only after target enrichment, which is more time-consuming [2, 9]. NGS directly from DNA or RNA extract without PCR amplification, as was done in the present case, is simpler and faster and allows simultaneous detection of several non-targeted infectious agents. Furthermore, NGS using ONT GridION allows viral genome detection and characterization within a timeframe that is appropriate for clinical diagnostic purposes. Thus, although still in its infancy, with necessary improvements in sensitivity and precision, ONT should optimize the diagnosis of infections in the near future.
Go to : 
 XML Download
XML Download