

 PDF
PDF Citation
Citation Print
Print



Dear Editor,
Pathogenic variants of TTN have been reported to cause various myopathies, including hereditary myopathy with early respiratory failure (HMERF; OMIM 603689) [1]. As TTN is a large gene [2], its variants may cause diverse phenotypes. We report a family with a novel, likely pathogenic variant of TTN and mild clinical symptoms that manifested at old age.
This study was approved by the Institutional Review Board of Hanyang University Guri Hospital (2021-05-032) and informed consent was obtained from the patient. Experiments were conducted according to the Declaration of Helsinki. The proband (II-5), a 69-year-old male, was admitted to Hanyang University Guri Hospital, Guri, Korea, for vitrectomy in December 2019. He had hypertension and diabetes mellitus. He reported frequent arousal during sleep, without any respiratory symptoms. At admission, oxygen saturation was 85%, arterial pH was 7.28, arterial carbon dioxide partial pressure (PaCO2) was 74.6 mm Hg, and arterial oxygen partial pressure was 52.2 mm Hg. After the application of noninvasive ventilator support, arterial PaCO2 reached 48.3 mm Hg and pH was 7.35.
Bilateral diaphragm elevation was revealed by chest computed tomography (Fig. 1A). A pulmonary function test showed a severe restrictive pattern (Fig. 1B and 1C). Sniff test showed bilateral diaphragmatic paralysis (Fig. 1D and 1E). We observed a lack of movement on ultrasonography. Both echocardiography and electrocardiography monitoring were normal.
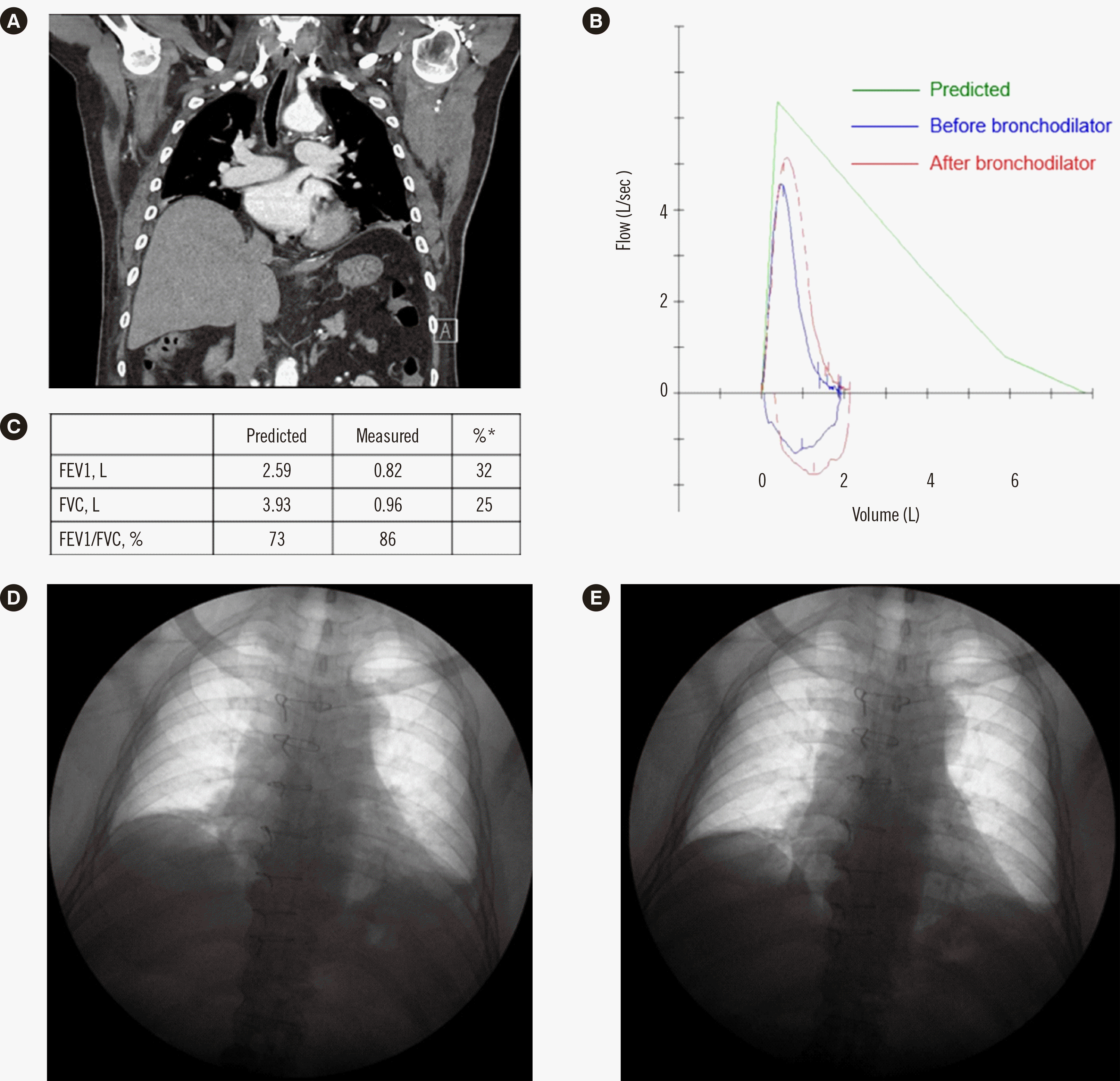 | Fig. 1Chest images and results of pulmonary and diaphragm function tests. (A) Chest computed tomography of the proband revealed bilateral diaphragm elevation. (B, C) Pulmonary function test showed a severe restrictive pattern, with negative bronchodilator test result. Chest X-ray was performed at full inspiration (D) and full expiration (E). Sniff test showed bilateral diaphragmatic paralysis. *Percentage of measured value compared with predicted value.
Abbreviations: FEV1, forced expiratory volume in one second; FVC, forced vital capacity.
|
Six months later, the proband reported limb-girdle muscle weakness, but manual muscle strength was nearly normal (Medical Research Council grade 5). We found no hypertrophy or atrophy. Serum creatine kinase level was slightly elevated (3.98 μkat/L). Nerve conduction was normal. Denervation potential was noted in thoracic paraspinal muscles and the right tibialis posterior on electromyography, but we detected no myopathic abnormalities.
The proband’s 74-year-old sister (II-7) reported similar symptoms (Fig. 2A), including slowly progressive proximal and respiratory muscle weakness. Similar to the proband, her symptoms manifested at old age. She showed no definite muscle wasting or weakness. Additional information could not be obtained. The proband’s mother (I-2) and brother (II-3) had similar clinical features, including respiratory failure, and they later died due to sudden cardiac arrest.
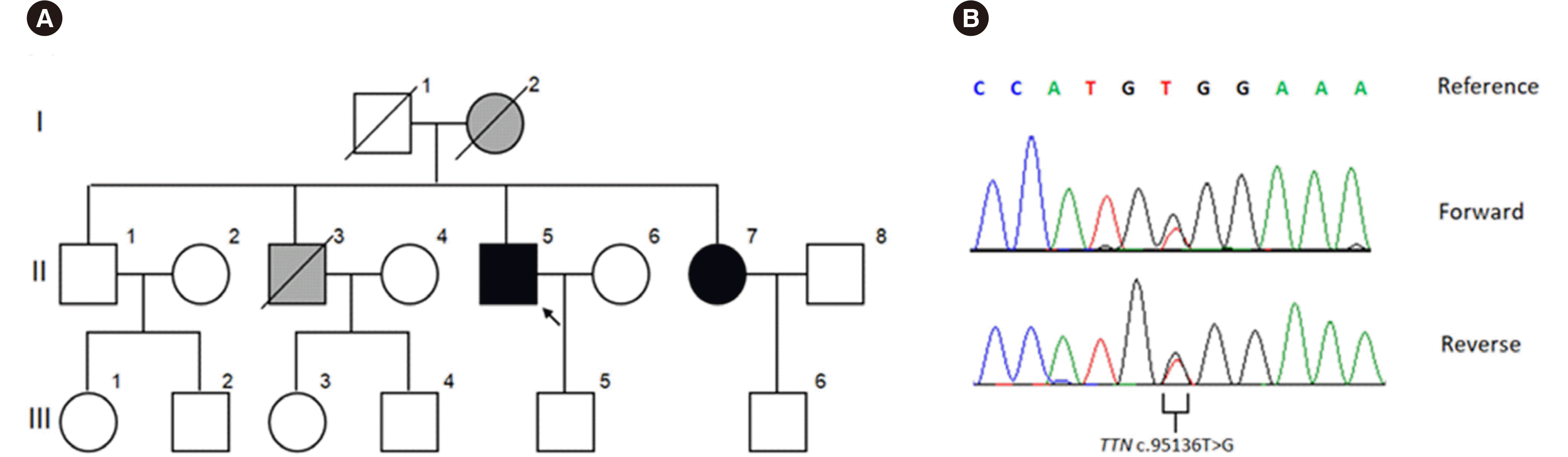 | Fig. 2Pedigree and sequence chromatogram of the novel TTN variant identified in the family. (A) Family pedigree. Open symbols indicate no signs or symptoms of hereditary myopathy with early respiratory failure (HMERF). Filled symbols represent affected individuals. Grey symbols indicate individuals who likely carry but were not tested for the TTN variant. The arrow indicates the proband. (B) Results of the likely pathogenic variant of TTN for the proband. Sequencing analysis revealed the NM_001267550.1:c.95136T>G (p.Cys31712Trp) (or NP_001243779.1:p.Cys30071Trp) variant.
|
Genomic DNA was extracted from peripheral blood of the proband and the proband’s sister.
Whole-exome sequencing for the proband’s sister at the Hospital of the University of Pennsylvania, Philadephia, USA, in January 2020 revealed a novel heterozygous NM_001267550.1:c.95136T>G (p.Cys31712Trp) (or NP_001243779.1:p.Cys30071Trp) TTN variant. Through Sanger sequencing, we found the same heterozygous TTN variant in the proband (Fig. 2B). The p.Cys31712Trp variant was absent in large population databases, i.e., gnomAD (https://gnomad.broadinstitute.org/), the Korean Reference Genome Database (http://coda.nih.go.kr/coda/KRGDB/), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), and the Human Gene Mutation Database Professional (version released in 2018; http://www.hgmd.org/), at the time of this study. The variant is located in the 119th fibronectin-3 domain (FN3 119) in the A-band region of TTN, where all HMERF-associated TTN pathogenic variants are located [3]. The p.Cys31712Trp variant is located at the same amino acid position as p.Cys31712Arg (or NP_001243779.1:p.Cys30071Arg), the most common variant in HMERF [4]. This variant was also predicted to be deleterious in in-silico analysis using SIFT (https://sift.bii.a-star.edu.sg/) and Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/). Based on our results, the TTN p.Cys31712Trp variant was considered a “likely pathogenic” variant, according to the 2015 American College of Medical Genetics and Genomics and the Association for Molecular Pathology guidelines [5, 6].
The relationships between individual pathogenic variants of TTN and different phenotypes have not been fully identified. Usually, disease onset due to pathogenic variants of TTN is from early to young adulthood and is characterized by respiratory failure or lower limb weakness [1, 7]. The clinical features of the patients in our study were compatible with HMERF [8]. The mean age of HMERF onset ranged from 16 to 53 years [7]. The degree and progression rate of muscle weakness are variable. However, to our knowledge, respiratory insufficiency at the age of 69 years without definite limb muscle atrophy or weakness has not been reported previously. Interestingly, the first symptom of HMERF in elderly patients may be respiratory failure caused by diaphragmatic paralysis.
The p.Cys31712Arg variant is the most common variant in HMERF and impairs proper folding of the FN3 119 domain [9]. The p.Cys31712Trp variant found in the current study may affect protein function and be related to a different phenotype. Titin is a giant structural muscle protein encoded by TTN [1, 8]. It comprises four structurally and functionally distinct regions that interact with numerous protein partners [10]. This implies that different pathogenic variants can have diverse biological effects, which may cause various phenotypes, including those seen in heterogeneous muscular disorders.
We reported patients with a mild form of HMERF that may be associated with a novel, heterozygous, likely pathogenic variant of TTN. Evaluation of TTN may be helpful in patients with suspected hereditary muscular disorder-related respiratory failure. Further studies are needed to confirm whether this variant is pathognomonic and to reveal relationships between clinical manifestations and this TTN variant.
Go to : 
 XML Download
XML Download