

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Mitochondrial DNA (mtDNA) depletion syndrome comprises a group of diseases caused by a deficiency of proteins involved in mtDNA synthesis, leading to quantitative (mtDNA depletion) and qualitative (multiple mtDNA deletions) defects in mtDNA.1 The diseases are classified based on the organs predominantly affected, such as encephalohepatopathy, encephalomyopathy, encephaloneuropathy, neurogastrointestinal encephalopathy, myopathy, ophthalmoplegia, optic atrophy, and neuropathy.2 MPV17 is located on chromosome 2p23-21 and contains eight exons. MPV17 is an mtDNA maintenance protein whose role remains to be elucidated. MPV17-deficient mice exhibit an insufficiency of mitochondrial deoxynucleotides, especially in the liver.3 MPV17-related hepatocerebral mtDNA depletion syndrome (also called an MPV17-related mitochondrial DNA maintenance defect) was first reported in 2006 and is an autosomal recessive disease characterized by encephalohepatopathy.4 Until 2018, approximately 100 individuals had been diagnosed with MPV17-related hepatocerebral mtDNA depletion syndrome. Most were described as an early-onset hepatocerebral disease. Late-onset neuromyopathic disease associated with multiple mtDNA deletions in muscles has rarely been described.5 This case report describes the clinical manifestations of MPV17-related hepatocerebral mtDNA depletion syndrome in a Korean child by whole-exome sequencing (WES).
CASE REPORT
A 17-month-old Korean girl was admitted to Seoul National University Children Hospital to understand her developmental delay and failure to thrive. She was born as a dichorionic diamniotic twin infant to nonconsanguineous parents at 37+1 weeks of gestation with a birth weight of 2.04 kg (<3rd percentile), height of 45.6 cm (50th percentile), and head circumference of 32.0 cm (50th percentile). Immediately after birth, she was admitted to a nursery for routine neonatal care. The liver function (AST, ALT, total bilirubin) and newborn screening test results were normal.
Her vital signs were normal during admission. Her height, weight, and head circumference were 70.6 cm (<3rd percentile), 7 kg (<3rd percentile), and 41.4 cm (<3rd percentile), respectively. Her motor developmental age was 10 months, as assessed using Gross Motor Function Measure with a score of 63.07%. A physical examination revealed central hypotonia and hepatomegaly. The laboratory tests showed elevated transaminases, ALP, GGT (AST 330 U/L, ALT 150 U/L, ALP 462 U/L, GGT 81 U/L), lactic acidemia (lactic acid 4.5 mmol/L, reference range 0.8-1.5 mmol/L), hyperbilirubinemia (total bilirubin 4.7 mg/dL, reference range 0.2-1.2 mg/dL; direct bilirubin 4.18 mg/dL, reference range 0-0.5 mg/dL), and coagulopathy (PT INR 1.87, reference range 0.8-1.2). The lactic acid-to-pyruvate ratio was 45.04. The newborn screening test and serology, including hepatitis A virus, hepatitis B virus, hepatitis C virus, herpes simplex virus, Epstein-Barr virus, and cytomegalovirus, were unremarkable.
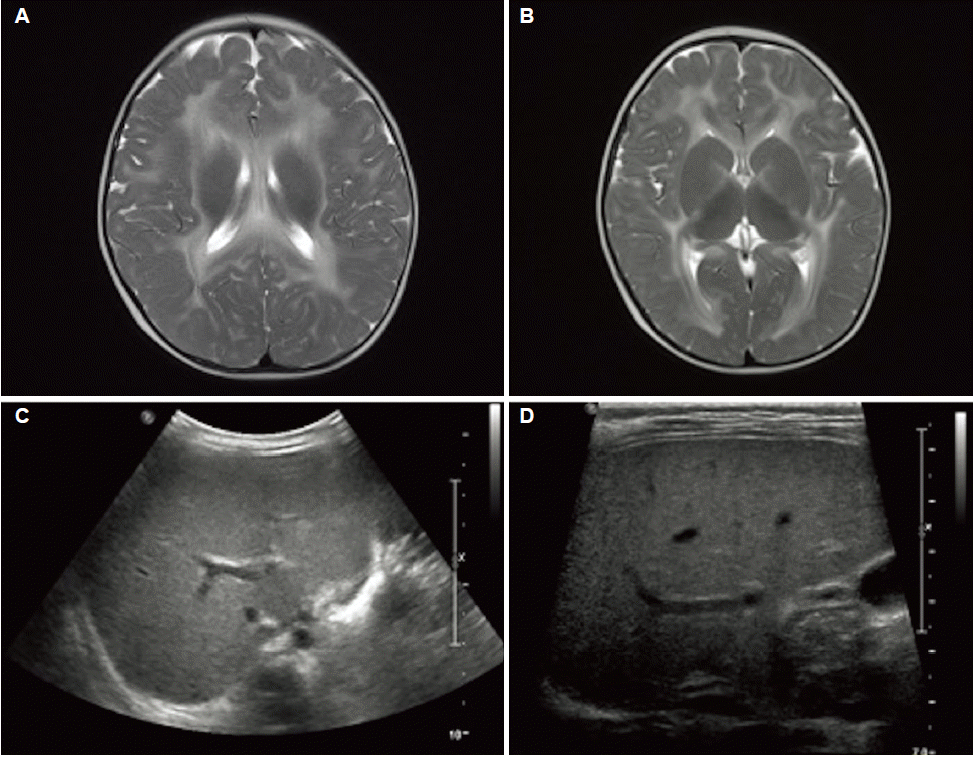
The abdominal ultrasound showed increased parenchymal echogenicity and periportal edema, and elastography revealed increased liver stiffness. Brain MRI showed extensive white matter T2-hyperintensity, suggesting a hypo-state or demyelination state (Fig. 1). Thus, the patient received fat-soluble vitamin replacement and medium-chain triglyceride supplementation. Subsequently, her total bilirubin and direct bilirubin levels decreased to 1.1 mg/dL and 1.02 mg/dL, respectively.
She was infected with adenovirus that manifested as diarrhea, vomiting, fever, and poor oral intake. She visited an emergency room because of a seizure. Her initial blood sugar level was 11 mg/dL; thus, she was fed uncooked cornstarch.
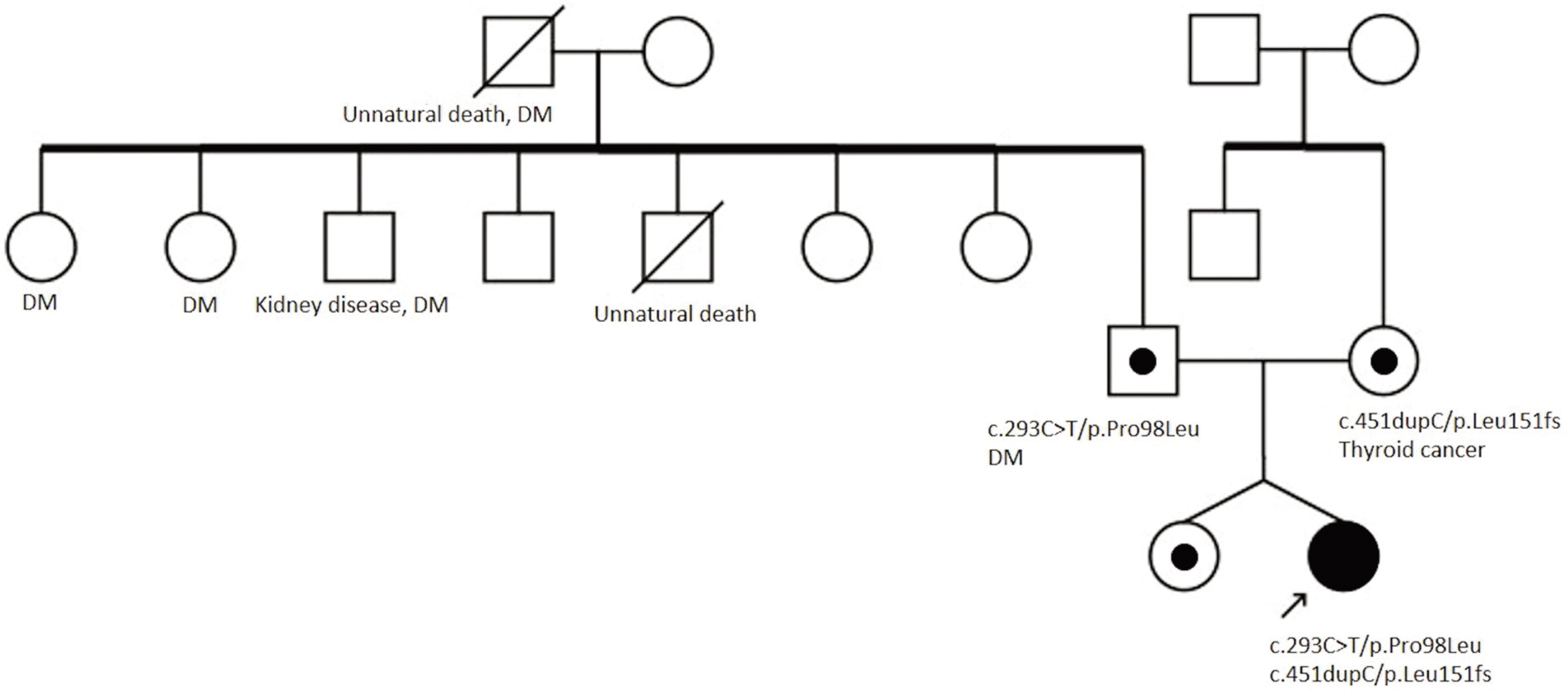
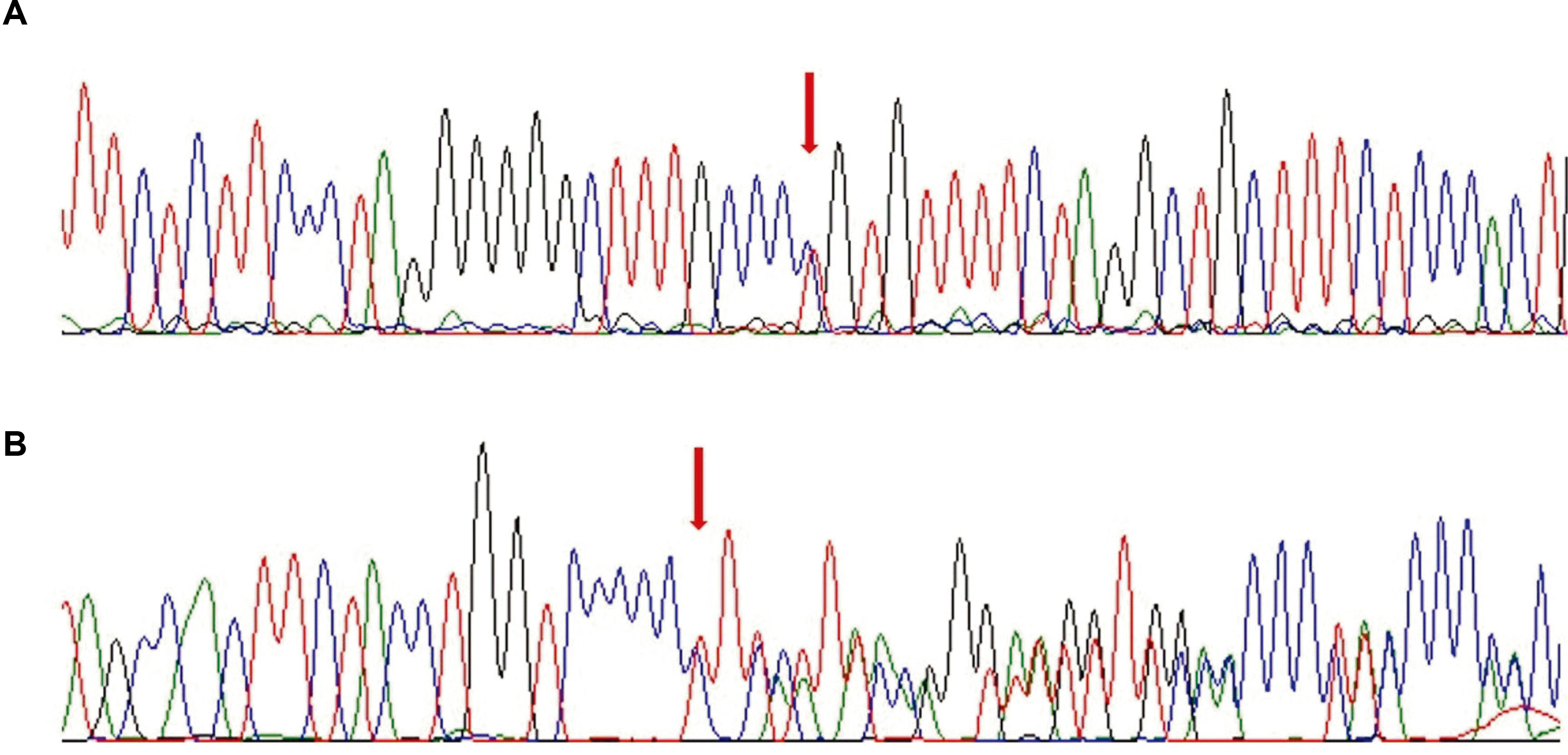
WES was performed using the genomic DNA from the peripheral leukocytes, and mutations were detected in MPV17. The patient had compound heterozygous c.451dupC/p.Leu151fs and c.293C>T/p.Pro98Leu mutations. Family screening revealed that her mother and father were heterozygous carriers of p.Leu151fs and p.Pro98Leu in MPV17, respectively. Her sibling was also a heterozygous carrier (Fig. 2). The mutations were confirmed by Sanger sequencing (Fig. 3).
Although the patient exhibited developmental delay, she underwent physical therapy for eight months and showed improvement in development. Her gross motor function measure score was 86.9%, but her liver function tests were aggravated. Despite the administration of vitamin K, her serum total bilirubin level was 2.6 mg/dL (direct bilirubin level 2.27 mg/dL) and PT INR was 3.21. Subsequently, she manifested with liver failure and mild neurological symptoms and is being considered for deceased donor liver transplantation. The Institutional Review Board of Seoul National University Hospital approved this study (Approval No. 1807-137-961).
DISCUSSION
Although MPV17-related hepatocerebral mtDNA depletion syndrome is a rare disease, its prevalence is unknown. Hepatic and neurological manifestations are substantial features of the disease and show early onset during the neonatal period or infancy. The patient presented with cholestatic hepatitis, failure to thrive, developmental delay, generalized hypotonia, hypoglycemia, and lactic acidemia. These clinical features and supportive laboratory findings were similar to those reported for MPV17-related hepatocerebral mtDNA depletion syndrome.5 Typical brain MRI scans show hypomyelination, leukodystrophy, and signal abnormality in the brain stem or basal ganglia.5,6 MPV17-related hepatocerebral mtDNA depletion syndrome is associated with a poor prognosis due to the progression of liver failure. In Korea, all cases of MPV17-related hepatocerebral mtDNA depletion syndrome died of liver failure.7
The potential of WES was introduced in 2009,8,9 and has since been improved. WES is a cost-effective and useful genetic diagnostic tool.10,11 In addition to MPV17, mutations in DGUOK, POLG, TFAM, and TWNK cause hepatocerebral mtDNA depletion syndrome.1 WES allows researchers to perform multiple genetic analyses for hepatocerebral mtDNA depletion syndrome. In particular, when the patient’s phenotype is analyzed, it is important to consider other congenital disorders, such as mitochondrial disease or disorder of glycosylation, in addition to mitochondrial DNA depletion syndrome. Compared to other molecular genetic tests, WES has strength in effectiveness when multiple genetic diseases are considered in a differential diagnosis. Thus, WES was performed to confirm the compound heterozygous mutations in MPV17. Hence, diagnostic examinations, such as liver biopsy and other genetic studies, were not necessary. WES can provide information on the prognosis, genetic counseling, and prenatal diagnosis.
To date, 48 MPV17 mutations have been reported. p.Gln93Pro is the most common homozygous pathogenic variant found in multiple Arab families.5 p.Leu151fs and p.Pro98Leu variants have also been detected in other cases. A recent study revealed the presence of p.Leu151fs in eight out of 13 Japanese patients (61.5%) with MPV17-related hepatocerebral mitochondrial DNA depletion syndrome.12 Thus far, 13 cases of p.Pro98Leu have been reported, among which seven patients (53.8%) were Asian.5,12
Genotype-phenotype correlations caused by MPV17 gene mutations are unclear. On the other hand, a trend of survival was observed depending on the variant type. Mortality is slightly lower in biallelic missense pathogenic variants than biallelic null (nonsense, frameshift, deletions, and slice-site) variants. Furthermore, individuals homozygous for p.Arg50Gln, p.Pro98Leu, or p.Arg41Gln may have a better prognosis.5
Previously, several MPV17 gene mutations were reported in Korea. Kim et al.7 described four Korean children with hepatoencephalopathy. A novel mutation, p.Val66Glu, was reported. Choi et al.13 encountered two cases of late-onset polyneuropathy without hepatoencephalopathy. They reported a novel homozygous mutation, p.Arg41Glu.
The outcome of liver transplantation (LT) in patients with MPV17-related hepatocerebral mtDNA depletion syndrome remains unclear because of its extrahepatic manifestations and poor outcomes during the post-transplantation period. The causes of death after LT are sepsis, respiratory failure, or multiorgan failure. Moreover, several patients developed pulmonary hypertension after LT.12 Nine out of 20 patients (45%) survived after LT.5,12,14 On the other hand, a case with the same mutation as the present patient (p.Leu151fs and p.Pro98Leu) showed a good prognosis with survival for more than 20 years after LT, even though the patient experienced neurological deterioration.12 Collectively, patients harboring p.Pro98Leu or p.Pro98Leu in at least one allele without marked neurological manifestations might have a better prognosis after LT.12 The patient with mild neurological manifestations is being considered for a deceased donor LT owing to liver failure.
In conclusion, this paper reported the clinical manifestations of MPV17-related hepatocerebral mitochondrial DNA depletion syndrome in a Korean girl. WES can be useful in diagnosing patients with hepatopathy, developmental delay, lactic acidosis, and hypomyelination on brain MRI.
 XML Download
XML Download