

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Weiss-Kruszka syndrome (WSKA; MIM#618,619) is a rare autosomal dominant disorder associated with zinc-finger protein 462 (ZNF462) gene variants or the deletion of the 9q31.2 chromosome region involving ZNF462. It is characterized by mild global developmental delay, ptosis, dysmorphic craniofacial abnormalities including metopic ridging or synostosis and triangular shape forehead with/without autistic features.12 The ZNF462 gene encodes a zinc-finger protein of unknown function3 and it was reported that its heterozygous loss-of-function variants presented with a pattern of overlapping phenotype in eight individuals from six families in 2017.4 To date, only 25 affected individuals from 22 families have been described.12 In most cases (95%), WSKA occurs due to de novo variants in ZNF462, and only 5% of individuals diagnosed with WSKA have an affected parent.1 Brain lesions related to WSKA are identified as ventriculomegaly and midline structural abnormalities of brain such as corpus callosum hypoplasia in about 25% of patients.2 However, there has been no report of structural malformation of the pituitary gland related to midline brain structures and endocrine dysfunction in individuals with WSKA. Here, we present the case of a Korean boy with molecularly confirmed WSKA and previously unreported clinical manifestations of primary empty sella syndrome (ESS), which was associated with growth hormone deficiency (GHD).
CASE DESCRIPTION
A 16-year-old boy was referred to our hospital for the evaluation of growth retardation and delayed puberty. He was born at full term to healthy and non-consanguineous Korean parents, with a birth weight and length of 3.0 kg (−0.30 standard deviation score [SDS]) and 50 cm (0.06 SDS), respectively. The heights of his father and mother were 174 cm (−0.08 SDS) and 170 cm (1.74 SDS). At the first visit, his height was 152.8 cm (−3.49 SDS) and weight was 42.7 kg (−2.82 SDS). There were no postnatal problems, such as microphallus and cryptorchidism, except bilateral ptosis. He underwent surgery for the correction of ptosis at the age of 4 years. He had no previous history of trauma or chronic illness. He complained of mild hypotonia when he exercised or did hard work. He felt frequent tiredness and muscle weakness in ordinary life. According to his mother, he worked alone at 15 months-old and his language development was also delayed when he was a young child. According to the results of his cognitive and intellectual function in the department of psychiatry at 18 years old, the full scale intelligence quotient was 75 according to the results of Korean Wechsler Adult Intelligence Scale-IV administered at the age of 18. However, the social age for estimating social adaptation function was social quotient 84, and considering the stable subtest performance level, his intellectual ability was estimated to be at the lower average level (80–89). In the comprehensive attention test, a decrease in the function of interference in selective attention and divided attention was observed.
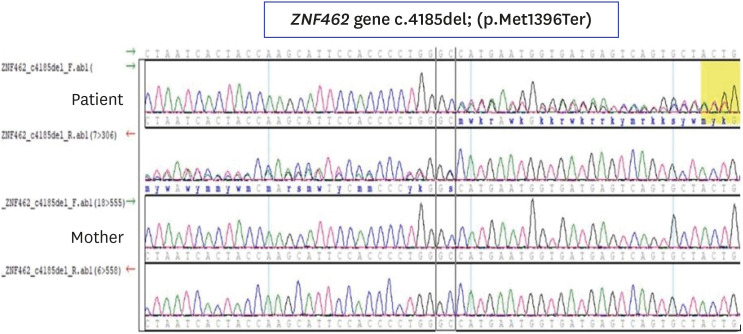
From school age, he gradually showed a decrease in height velocity, and there has been no pubertal development in adolescence. He had two younger brothers who grew up normally without any growth and developmental issues and had no family history of genetic disorders. The blood pressure was 109/73 mmHg. His pubertal stage was assessed as Tanner 2 for the external genitalia development (testes volume 4 mL). He had dysmorphic craniofacial features, including an inverted triangular-shaped forehead, exaggerated Cupid's bow, arched eyebrows, down-slanting palpebral fissures, pterygium in the left eye with corneal opacity, mild ptosis and poorly expressive face (Fig. 1). He presented with mild hypotonia without other neurological symptoms and signs such as headache or visual disturbance. Laboratory tests for chemistry and thyroid hormone showed normal levels. The serum insulin-like growth factor (IGF)-1 and IGF binding protein-3 levels were 155.3 ng/mL (normal range: 360.0–885.0 ng/mL) and 1,698 ng/mL (normal range: 1,574–4,260 ng/mL), respectively. The bone age was over 2 years delayed. Two different provocative growth hormone (GH) testing, glucagon stimulation test and L-dopa stimulation test, revealed complete GHD as the result of low peak GH levels (0.34 and 1.99 ng/mL, respectively). Gonadotropin-releasing hormone stimulation testing showed early stage of puberty (peak luteinizing hormone 8.93 mlU/mL and peak follicle stimulating hormone 12.10 mIU/mL). Testosterone level was 0.23 ng/mL. Magnetic resonance imaging (MRI) of the brain showed a flattened pituitary gland and cerebrospinal fluid (CSF) space herniated into the sella turcica, which indicated ESS (Fig. 2). The other pituitary hormones showed normal levels. To identify the underlying genetic cause, we performed karyotype and chromosomal microarray analysis, which showed unremarkable results. Whole exome sequencing (WES) was performed and a heterozygous novel nonsense variant, c.4185del; p.(Met1396Ter), in ZNF462 was identified, which was confirmed by Sanger sequencing (Fig. 3). This variant resulted from the deletion of nucleotides “C” in exon 3 of ZNF462, making stop codon in the protein-coding sequence, which was expected to produce truncated protein. This variant in ZNF462 has not been reported in a large population database as well as in the Genome Aggregation Database (https://gnomad.broadinstitute.org/). The patient's phenotype matched the typical phenotypes of previously reported individuals with WSKA.5 Segregation analysis using Sanger sequencing showed no ZNF462 variant in his mother. Although his father and two younger brothers could not undergo genetic tests for detecting the ZNF462 variant, there were no clinically affected family members. Therefore, this variant was categorized as pathogenic according to the standards and guidelines of the American College of Medical Genetics and Genomics.6 There were no additional congenital anomalies such as congenital heart defects, optic nerve hypoplasia, papilledema and hearing impairment. Recombinant human growth hormone (rhGH) replacement therapy was started at the dose of 0.23 mg/kg/week administered as daily subcutaneous injections and the dose of rhGH has been gradually increased up to 0.3 mg/kg/week until 18 years old. An improvement in height velocity (8 cm/year) was observed and his current height is 168.2cm (−1.15 SDS). Gradual pubertal progress (Tanner stage 2 for the external genitalia development, testes volume 6 mL at 18 years old) was also observed (Fig. 4). Now, he is 18 years 11 months-old and his bone age was estimated at 15 years. He continues rhGH replacement therapy and we consulted a urologist to evaluate and manage sexual maturity and fertility.
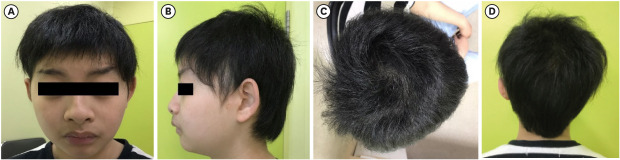
Fig. 1
Patient's craniofacial manifestation. (A, B) The patient has arched eyebrows, exaggerated Cupid's bow, and short upturned nose with bulbous nasal tip. (C) Inverted triangular-shaped forehead was shown. (D) Inverted triangular-shaped head shape was shown in back. The figures are published with the consent of the patient and his parents.
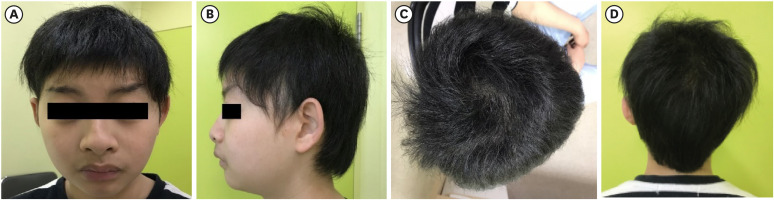
Fig. 2
MRI studies of the brain showing ESS. (A) Sagittal view of brain MRI shows a flattened pituitary gland and CSF space herniated into the sellar turcica. The posterior pituitary gland appears normal. (B) Coronal view of brain MRI shows the pituitary fossa which is largely empty of tissue, replaced by CSF. (C) Axial view of brain MRI shows ESS.
MRI = magnetic resonance imaging, ESS = empty sella syndrome, CSF = cerebrospinal fluid.
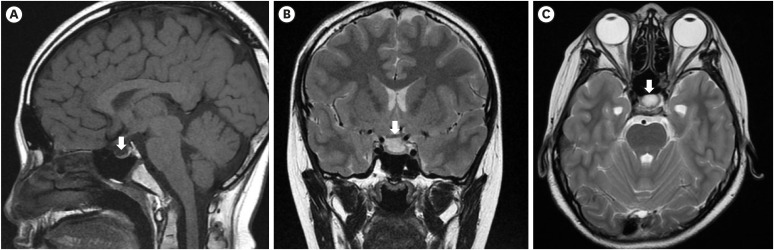
Fig. 3
Growth chart of the patient under rhGH replacement therapy. Upper side shows height chart and lower side shows weight chart. The dots indicate the height and weight measurement. The arrow marks the beginning of rhGH treatment start. The patient's height velocity after the start of rhGH was increased 8 cm/year.
rhGH = recombinant human growth hormone.
DISCUSSION
ZNF462 located in the 9q31.2 chromosome encodes a C2H2 type of zinc-finger transcription factor, which is believed to play an important role in embryonic development, especially in brain development, and it controls early patterning of the central nervous system.2789 According to the literature, the phenotype associated with variations in ZNF462 is characterized by craniofacial anomalies, corpus callosum dysgenesis, ptosis, and developmental delay.47 Since the first reported case of a ZNF462-related syndrome in 2003,128 only 25 individuals from 22 families have been identified to have pathogenic variants in ZNF462, including chromosomal rearrangements disrupting ZNF462.127
The most common clinical features of WSKA are ptosis and developmental delay, described in 83% and more than 75% of affected individuals, respectively.12 Patients with WSKA show various types of developmental delay, such as global delay, motor delay, speech delay, or a combination of these. In addition, down-slanting palpebral fissures (54%), an exaggerated Cupid's bow (54%), arched eyebrows (50%), epicanthal folds (46%), and a short, upturned nose with a bulbous tip (46%) have been described in a large proportion of patients.12 However, metopic ridging or craniosynostosis involving the metopic or lambdoid suture is less common (38%).128 About 25% of patients with WSKA show corpus callosum abnormalities on brain imaging (Table 1).12 Our patient had typical craniofacial features, which were previously reported in individuals with WSKA, including bilateral ptosis, mild intellectual disability without autism, and mild hypotonia. Interestingly, after the diagnosis of WSKA based on the results of WES, “reverse phenotyping” was performed to confirm the diagnosis. As previously reported, the extensive use of facial analysis technologies can help increase the number of patients diagnosed with this syndrome.12 Additionally, brain MRI of this patient indicated ESS, which is associated with hypothalamic-pituitary dysfunction, such as complete GHD; this clinical finding was unique and has not been reported. The index patient in our study carried a novel pathogenic heterozygous nonsense variant, c.4185del; p.(Met1396Ter), in exon 3 of the ZNF462 gene. Most of the reported pathogenic variants in ZNF462 are loss-of-function variants in exon 3, which accounts for 54% of the coding region of ZNF462.8 Another heterozygous variant in exon 3 of the ZNF462 gene, c.4165C>T; p.(Gln1389Ter) (NM_021224.5), has already been reported; it is located in close proximity to our patient's variant position.8 A patient with a Gln1389-to-Ter substitution showed grossly similar craniofacial features with no mention of growth failure and he did not undergo brain MRI.8
Table 1
Phenotypes of patients with reported variants of ZNF462
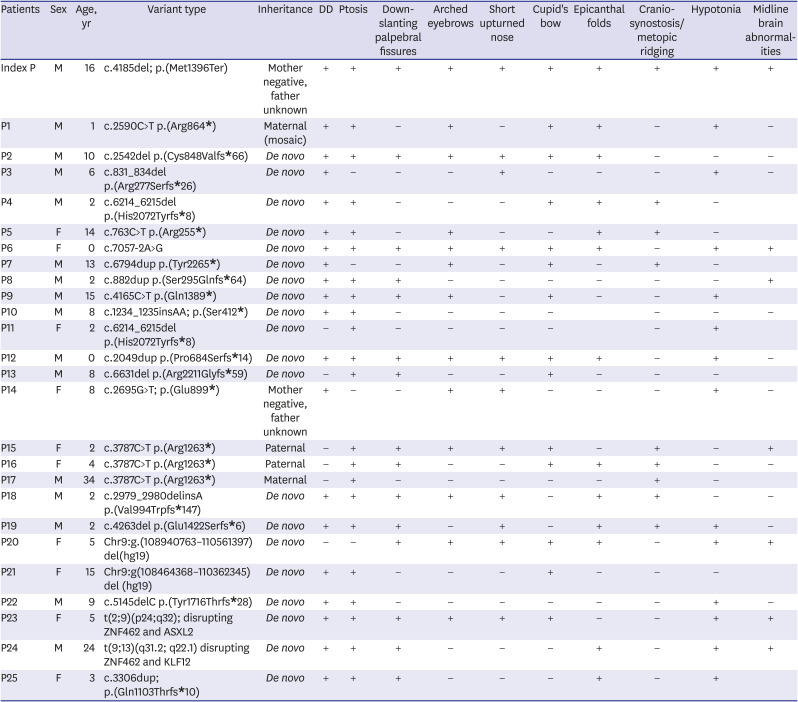
Inheritance types were maternal 8% (2/25), paternal 12% (3/25), unknown 8% (2/25), de novo 72% (18/25). Clinical characteristics were below; DD in 76% (19/25), 84% (21/25) with ptosis, 56% (14/25) with down-slanting palpebral fissures, 52% (13/25) with arched eyebrows, short upturned nose in 48% (12/25), Cupid's bow in 52% (13/25), metopic ridging in 40% (10/25), hypotonia in 52% (13/25) and midline brain abnormalities in 28% (7/25, 10 had normal brain MRI findings and 8 were not tested) including our patient. Blank means no mention about the clinical features and/or no test results have been reported.
P = patient, DD = developmental delay, MRI = magnetic resonance imaging.
Primary ESS is observed less frequently in children than in adults, and it is frequently associated with hypothalamic-pituitary dysfunction such as GHD, hypogonadism, and multiple pituitary hormone deficiency.101112 The exact etiology of primary ESS is not known, but several etiopathogenetic hypotheses have been proposed, including congenital deformities of the sellar diaphragm and pituitary and/or upper sellar factors.1013 The following syndromes have been reported to be associated with primary ESS: Turner syndrome, Moyamoya disease, Bartter's syndrome, nevoid basal cell carcinoma syndrome, Hunter syndrome, Prader-Willi syndrome, Alstrom syndrome, Meniere's disease, and Erdheim-Chester disease.13 To date, a few patients with WSKA who underwent brain imaging (64%, 16/25) have shown corpus callosum dysgenesis (30%, 5/16).2 Since the causative gene for this syndrome has recently been identified, its clinical manifestation and molecular basis need to be established simultaneously. However, considering that the pituitary gland, pons, cerebellar vermis and corpus callosum are included in brain midline structures, we infer ESS might be a type of deformity in brain midline structures.14 As ZNF462 significantly contributes to the regulation of brain morphogenesis,1516 further functional studies on ZNF462 variants are required.
This is the first case of WSKA accompanied by primary ESS associated with GHD. This case contributes to the diagnosis of WSKA and the identification of its clinical features. More clinical and functional studies are needed to elucidate this association.
 XML Download
XML Download