

 PDF
PDF Citation
Citation Print
Print



Adrenoleukodystrophy (ALD) is the most common peroxisomal disorder caused by mutations in the ATP-binding cassette protein subfamily D1 gene (ABCD1), which is located on the X chromosome. ABCD1 encodes for the peroxisomal membrane transporter ALD protein (ALDP), which is involved in the degradation of very-long-chain fatty acids (VLCFAs).1 Defects in ALDP result in abnormal accumulation of VLCFA in the nervous system, adrenal glands, and testes, causing various clinical manifestations.2 ALD is traditionally categorized into four phenotypes: cerebral ALD, adrenomyeloneuropathy (AMN), Addison’s disease only, and asymptomatic carriers.3 AMN usually manifests between the ages of 20 and 50 years with progressive noninflammatory axonopathy that primarily involves the spinal cord and peripheral nerves, leading to spastic paraparesis and sensory ataxia. Furthermore, inflammatory cerebral demyelination occurs in up to 60% of male patients.4 We report here a patient with AMN with cerebral involvement due to a novel frameshift variant in exon 1 of ABCD1.
CASE
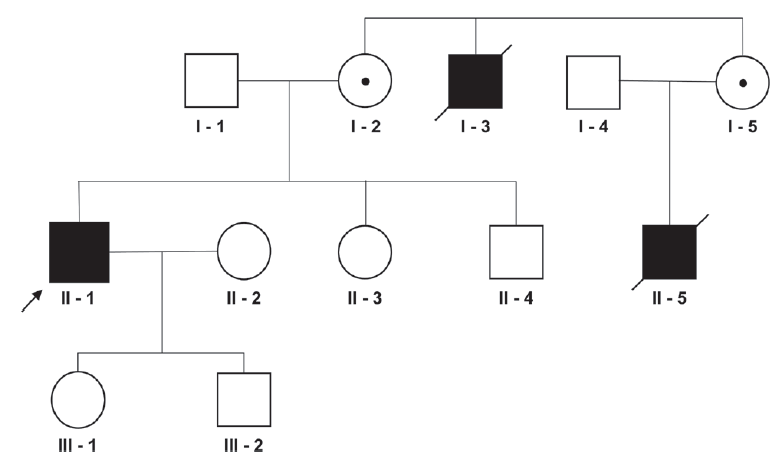
A 47-year-old male visited the outpatient clinic complaining of cognitive impairment, which had deteriorated over a period of 3 years. During his late 20s, the patient suffered from back pain and gait disturbance, which had slowly progressed. By his late 30s, he noticed a darkening of his skin and was taking sildenafil to treat erectile dysfunction. Over the past 3 years he experienced occasional falls due to a deterioration of his gait disturbance and had difficulties making appropriate decisions, finding directions, and making calculations. He reported no concurrent disease or relevant past medical history, including hypertension and diabetes. The patient did not have any history of intoxication or infection. However, he had a family history of cognitive impairment and gait disturbance, suggesting X-linked recessive inheritance (Fig. 1).
A neurological examination revealed an alert mental status, although disorientation for place was observed. He had a decreased Mini Mental State Examination score of 20. Cortical dysfunction was observed in multiple domains, including perseveration, ideomotor apraxia, acalculia, and finger agnosia. Cranial nerve examinations did not reveal any significant abnormalities. Motor power was intact, while deep tendon reflexes were hyperactive with positive bilateral Babinski and Hoffman signs. Bilateral limb ataxia was observed in the arms and legs, along with severely impaired position and vibration sense of lower extremities. His gait was wide-based, spastic, and ataxic.
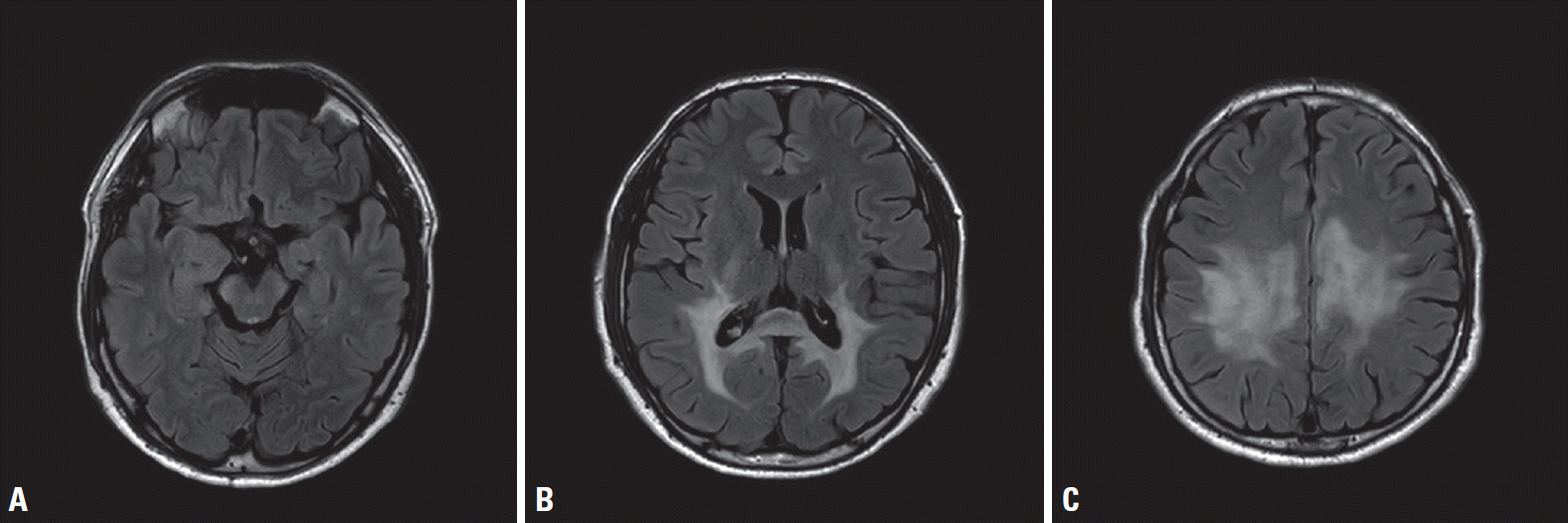
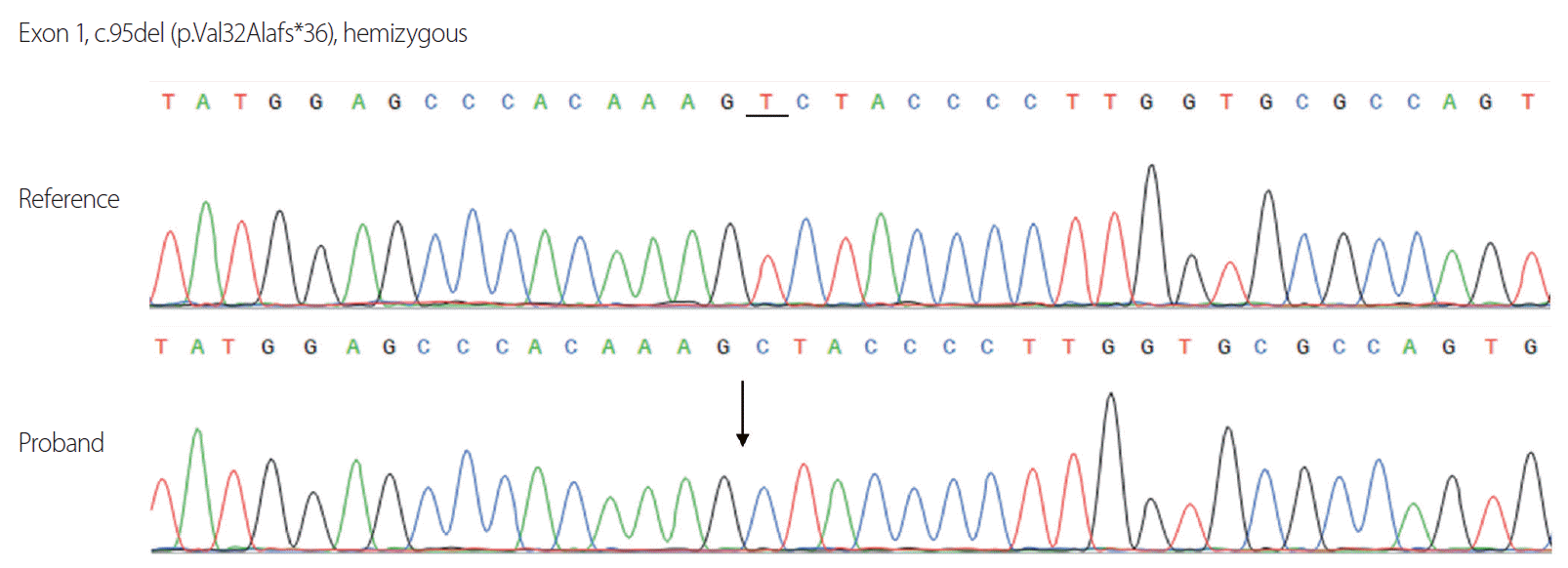
The baseline adrenocorticotropic hormone (ACTH) concentration measured in the morning was elevated at 161 pg/mL (reference 7-63 pg/mL), but the result of a rapid ACTH stimulation test was within normal limits (baseline serum cortisol level, 19 μg/dL; serum cortisol level at 60 minutes after stimulation, 27.4 μg/dL). Plasma VLCFA levels of C26:0, and C24:0/C22:0 and C26:0/C22:0 ratios were increased, at 3.8 μmol/L (reference ≤ 1.3 μmol/L), 2.0 (reference ≤ 1.39), and 0.093 (reference ≤ 0.023), respectively. Motor evoked potentials and posterior tibial somatosensory evoked potentials demonstrated central conduction delay, but a nerve conduction study did not reveal definitive electrophysiological evidence of neuropathy. Brain magnetic resonance imaging revealed increased signal intensities on T2-weighted images in the bilateral corticospinal tracts, the frontoparietooccipital subcortex, and the splenium of the corpus callosum (Fig. 2). Sanger sequencing of all 10 exons and flanking intron regions of ABCD1 (NM_000033.4) revealed a novel frameshift variant (c.95del [p.Val32Alafs*36]) in exon 1 (Fig. 3). This variant, which has not been reported previously in either ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) or the ALD database (http://www.x-ald.nl/), was classified as pathogenic based on the American College of Medical Genetics and Genomics standards, as follows: 1) null variant in a gene where the loss of function is a known disease mechanism, 2) absent from controls in the 1,000 Genomes Project (https://www.internationalgenome.org/) and gnomAD database (https://gnomad.broadinstitute.org/), and 3) in-silico analyses using MutationTaster (http://www.mutationtaster.org/) and LoFtool (https://asia.ensembl.org/Tools/VEP) supporting a deleterious effect.
DISCUSSION
We report here a case of AMN with cerebral involvement due to a novel frameshift variant that has not been reported previously. The phenotype of ALD is heterogeneous, but can be classified mainly as either cerebral ALD or AMN. The pathologies of cerebral ALD and AMN are fundamentally different; while the pathology of cerebral ALD is considered to be inflammatory demyelination, that of AMN is thought to be chronic neurodegeneration.5 In addition, the absence of a genotype-phenotype correlation has been demonstrated by several case series.4 These data support the hypothesis that Fig. 1. Pedigree of the patient. The arrow indicates the proband. a primary defect in ABCD1 results in AMN, but that additional environmental triggers or genetic modifiers are required to initiate cerebral demyelination.4
In a previous report of 12 Korean AMN patients that confirmed complex clinical phenotypes, 2 patients harbored a frameshift variant that manifested as the spinocerebellar phenotype.6 By contrast, the frameshift variant in our case manifested as AMN with cerebral involvement.6 In the aforementioned study, two patients were classified as having AMN with cerebral demyelination; one of these patients had a missense variant and the other had a deletion variant.6 In addition, it has been reported that adult cerebral ALD tends to involve the frontal rather than the parietooccipital area of the brain,7 but our adult-onset case manifested predominately with parietooccipital dysfunction. The findings of this case also support the lack of a genotype–phenotype correlation in ALD.
Treatments for ALD differ according to the targeted symptoms. Glucocorticoid supplementation is needed for adrenal insufficiency, and symptomatic management with supportive care is required for myelopathy. For cerebral ALD, allogeneic hematopoietic stem cell transplantation (HSCT) has been shown to halt disease progression in both childhood and adulthood cases.8 However, allogeneic HSCT has several risks, including treatment-related mortality, graft failure, graft-versus-host disease, and opportunistic infections.8 In 2017, a gene-therapy based approach was developed for the treatment of childhood ALD: autologous HSCT using autologous CD34+ stem cells corrected ex vivo with the lentiviral vector carrying wild-type ABCD1. This approach conferred long-term disease stabilization and had a reasonable safety profile.9 A gene therapy is also under development for AMN. Gong et al.10 found that delivery of an adenoviral vector containing human ABCD1 to mouse central nervous system cells resulted in localization of ALDP to the peroxisome, with a subsequent decrease in VLCFAs, both in vitro and in a mouse model of X-linked ALD.
Given the substantial progress in therapeutic interventions, the importance of accurate diagnosis is increasing. Clinicians should be aware of the heterogeneous clinical presentations and the lack of a genotype-phenotype correlation in ALD.
 XML Download
XML Download