

 PDF
PDF Citation
Citation Print
Print



Congenital myasthenic syndromes (CMSs) are rare genetic disorders characterized by weakness and fatigue resulting from impaired neuromuscular transmission. Although genetic testing can confirm the diagnosis for some types of CMS, variations in genotypes, clinical phenotypes, age at disease onset, and responses to treatment makes diagnosis very difficult.1 Here we present two adult patients who had significant decremental responses in repetitive nerve stimulation testing (RNST) and multi-minicore pathology, and who responded to treatment with a cholinesterase inhibitor.
CASE
A 40-year-old female was admitted due to slowly progressive muscle weakness and fatigue. A birth history could not be obtained because her parents died when she was young; however, she remembered was that she had been ‘weak’ and unable to run as fast as others since she was an elementary-school student. At the age of 35 years she underwent a caesarian section for her first baby, after which she had to depend on a mechanical ventilator due to respiratory failure after general anesthesia. Detailed medical records could not be obtained. The patient reported that her weakness and fatigue had subsequently worsened and she suffered from occasional respiratory difficulty and aspiration pneumonia from that point. She had received a diagnosis of myasthenia gravis (MG) and muscular dystrophies at another hospital. RNST yielded positive results but no muscle biopsy had been conducted. The patient had been prescribed oral pyridostigmine and corticosteroid to treat MG. Oral pyridostigmine slightly improved her symptoms, but neither the corticosteroid nor intravenous immunoglobulin (IVIg) was effective.
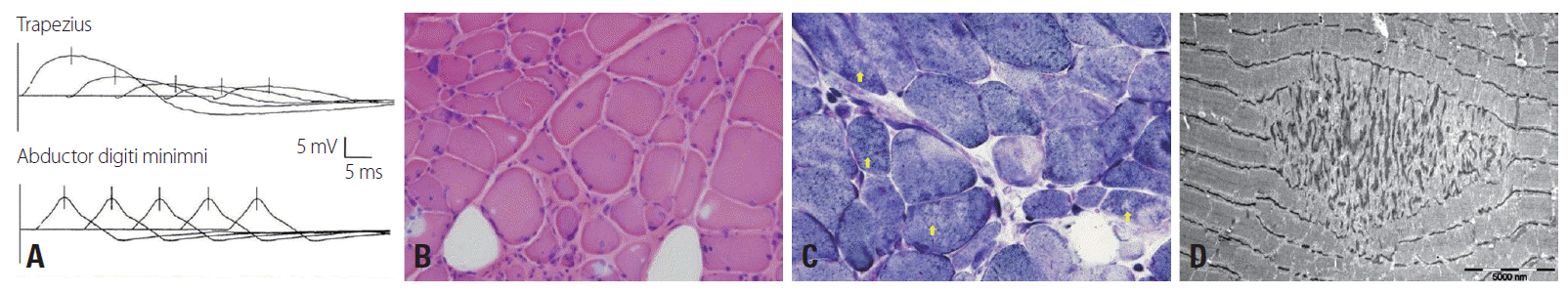
The patient had a low muscle mass and scoliosis. A neurologic examination revealed normal extraocular and facial muscles with respect to range of movement and strength, but mild ptosis. The muscle strength of the shoulder and hip girdle muscles was Medical Research Council grade 4-bilaterally, and grip power was normal. Sensory examination was normal, but deep tendon reflexes were absent in both the upper and lower extremities. Her serum creatine kinase level was slightly elevated at 256 IU (normal range, 32-135 IU/L) and she was negative for acetylcholine receptor antibodies (AChR-Ab). Motor and sensory nerve conduction studies produced normal findings. Electromyography performed for the right biceps, first dorsal interossei, vastus lateralis, and tibialis anterior muscles showed myopathic changes with mild fibrillation potentials. Significant decremental responses were noted in the right flexor carpi ulnaris (14.7%) and trapezius muscles (71.8%), but not in the abductor digiti quinti and facial muscles (Fig. 1A). The history of therapeutic failure of IVIg and corticosteroid raised the possibility of a diagnosis of MG, justifying a muscle biopsy. The muscle biopsy specimen from the left biceps muscle demonstrated increased variation of fiber size and connective tissues (Fig. 1B), multiple cores within the muscle fibers, and focal disintegration of myofibrillar structures (Fig. 1C, D). Immunohistochemical staining for dystrophin was normal. There were no pathologic features suggesting dystrophy or inflammatory myopathies.
We also examined her only brother, who was asymptomatic, and found generalized muscle atrophy, mild weakness of the proximal and axial muscles, and scoliosis. His serum creatine kinase level was slightly elevated at 234 IU, and RNST revealed a significant decremental response (24.5%) in the right trapezius muscle. Histology of a biopsy sample of the biceps muscle revealed multiple small zones of sarcomeric disorganization and muscle fibers lacking oxidative activity, consistent with multi-minicore disease (MmD).2 He showed modest clinical response only to the combination of pyridostigmine and 3,4-diaminopyridine.
DISCUSSION
The cases reported here presented proximal muscle weakness with relatively preserved distal limb and extraocular muscles, scoliosis, respiratory insufficiency, and significant decremental responses in RNST. The initial clinical diagnosis was AChR-Ab-negative limb-girdle MG. However, the lack of response to immunotherapy raised suspicion of another etiology, and muscle biopsy findings suggested MmD.
Our patient and her brother partially met the clinical diagnostic criteria of MmD. Although mutations in RYR1 and SEPN1 have been reported in approximately half of patients with MmD, most diagnoses are currently still based on a combination of clinical features and typical muscle pathologic findings.3,4 Indeed, multi-minicore pathology can be found in other conditions, including muscular dystrophy, inflammatory myopathies, and endocrine myopathy, but additional pathologic changes are also found in those diseases.5 Moreover, the obstetric history of respiratory insufficiency after general anesthesia for cesarean section in our patient was significant because it suggested that she might harbor a RYR1 mutation.
The significant decremental response in RNST appears to be unique to our patients. This decremental response in myopathies could result from multifocal disruption of the neuromuscular junction due to disintegration of the myofibrillar structures, and could explain the responsiveness of the patients to the cholinesterase inhibitor and 3,4-diaminopyridine.
It should be noted that CMSs cannot be ruled out; the variation in age at onset associated with CMSs and clinical similarities to MG means that CMSs may be easily mistaken for myopathy or seronegative MG.1 Patients with a CMS caused by mutations in DOK7 have a minimal decremental response in RNST, but multi-minicore-like pathology is often also revealed.6,7 However, prominent decremental responses in RNST and a good response to cholinesterase inhibitor treatment did not favor a diagnosis of a CMS with DOK7 mutation in our patients.
Since genetic testing was not available, we were unable to confirm whether the diagnosis was MmD or a CMS; however, we believe that these cases are worth reporting because they indicates that patients who are seronegative-MG but who exhibit atypical responses to standard treatments should always be checked for alternative diagnoses, including congenital diseases. Although MG and other congenital myasthenic myopathies coexist only rarely, their differential diagnosis is important because immunosuppressants are not helpful and may even do harm in patients with the latter. It would be helpful to take a meticulous medical history and perform extensive tests, including muscle biopsy and genetic analysis.
 XML Download
XML Download