

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Research and development of new therapeutics in neurology are expanding in an unprecedented manner. It is an exciting time for both clinicians and patients. Medical professionals must continually update their knowledge of these new drugs based on their mechanisms of action. The nomenclature is the road sign that guides this exploration.
International nonproprietary names (INN), commonly known as generic names, are unique identifiers of pharmaceutical substances.1 The World Health Organization (WHO) assigns these names in order to clearly identify medicines and facilitate the communication of information among health professionals and scientists worldwide. Many countries have councils to coordinate their naming system, such as British Approved Names, Dénominations Communes Françaises, Japanese Accepted Names, and United States Adopted Names (USAN).2 However, with rare exceptions, the nomenclatures adopted by these councils are identical to the INN as a result of ongoing collaboration.
Names of legacy drugs consist of two parts: a fantasy element assigned by the pharmaceutical company and a stem that reveals its class. For example, 3-hydroxy-3-methylglutaryl-coenzyme A inhibitors, which are indicated for hyperlipidemia, share the suffix ‘-vastatin,’ and beta blockers have the suffix ‘-alol.’ Some drugs have a second word that identifies its salt or ester, as in clopidogrel bisulfate or nortriptyline hydrochloride. We do not usually recognize the second word as an essential component, since it mainly reflects the drug’s physicochemical properties rather than the pharmacological action of the active pharmaceutical ingredient. Mycophenolate mofetil has a unique second word, which is the acronym of morpholino ethyl ester.
For a biological product, the United States Food and Drug Administration (FDA) requires the addition of an FDA-designated suffix in addition to the core name of the biological product. The function of this four-letter suffix is to distinguish the originator, related biological products, and biosimilar products, and it must be unique and devoid of meaning. For example, the biosimilar products of rituximab have rituximab-abbs (Truxima®, Celltrion, Incheon, Korea) and rituximab-pvvr (Ruxience®, Pfizer, New York, NY, USA). The proper names of the newer drugs carry this suffix from the originators, such as ravulizumab-cwvz (Ultomiris®; Boston, MA, USA).
Go to : 
PROTEIN THERAPEUTICS3
Parenterally administered purified protein can produce diverse pharmacological actions. The most famous in the field of neurology must be alteplase (Actilyse®; Boehringer Ingelheim, Ingelheim, Germany), more commonly known as tissue plasminogen activator. This therapeutic has revolutionized the treatment of cerebrovascular disorders. The suffix ‘-ase,’ as in urokinase, indicates that the active substance is an enzyme, in accordance with the biochemical nomenclature. The addition of ‘-tepl-’ ahead of ‘-ase’ specifically denotes that it is a tissue-type plasminogen activator, as in reteplase (Retavase®; Chiesi Farmaceutici, Parma, Italy)4 and tenecteplase (Metalyse®; Beohringer Ingelheim, Ingelheim, Germamy).
Several enzyme-replacement therapies have been introduced in the field of neurology. It is intuitive to add the suffix of ‘-ase’ to these drug names, since they are often the very enzymes that are deficient in the patients. Alglucosidase alfa (Myozyme® and Lumizyme®; Sanofi, Paris, France) is a recombinant acid alpha glucosidase (GAA) indicated for Pompe disease. It is the first specific therapy for the inherited myopathy. Avalglucosidase alfa is a second-generation recombinant GAA, which was chemically modified to carry synthetic bis-phosphorylated oligosaccharides to facilitate the mannose-6-phosphate receptor-mediated uptake process. The second word, alfa, is a Greek letter that identifies the product as the first glycoform of its kind. It is intentionally misspelled to comply with the phonetic rule that prefers f in place of ph.
Both agalsidase alfa (Replagal®; Takeda, Tokyo, Japan) and agalsidase beta (Fabrazyme® and Fabagal®; Sanofi, Paris, France) delivers alpha galactosidase (GLA), which is deficient in Fabry disease. The amino-acid sequence of these two drugs is identical, but they are dressed in different glycoforms, and are thus tagged with different Greek letters.5 The differences in their glycosylation patterns are attributable to the sources of their production: agalsidase alfa is produced in a genetically engineered continuous human cell line, while agalsidase beta is derived from Chinese hamster ovary (CHO) cells transduced with the gene encoding human GLA.
These Greek letter tails are reminiscent of older biological drug names familiar to neurologists. Both interferon beta-1a (Rebif®, Rebidose®, Merck, Kenilworth, NJ, USA; Avonex®; Biogen, Cambridge, MA, USA) and interferon beta-1b (Betaseron®, Bayer, Leverkusen, Germany; Betaferon®, Extavia®, Novartis, Basel, Switzerland) are human interferon beta 1, which is indicated for the treatment of multiple sclerosis. Interferon beta-1a is glycosylated as it is produced from CHO cells, while interferon beta-1b is purified from recombinant Escherichia coli and is not glycosylated. In addition, peginterferon beta-1a (Plegridy®; Biogen, Cambridge, MA, USA) is a form of interferon that has been pegylated in order to enable a reduced dosing frequency. Its name has the prefix ‘peg-’ derived from polyethylene glycol.
A unique suffix of ‘-mer’ in glatiramer (Copaxone®, Teva, Petah Tikva, Israel; Glatopa®, Novartis, Basel, Switzerland) announces that the drug is a polymer. The front part of its name indicates that it is a synthetic random polypeptide of glutamate, lysin, alanine, and tyrosine, with the composition of amino acids in myelin basic protein.6
Go to :
THERAPEUTIC MONOCLONAL ANTIBODIES7
Antibody drugs are a specialized group of protein therapeutics. The list of monoclonal antibody drugs has expanded exponentially with advances in antibody-engineering technologies. The names of monoclonal antibodies are composed of head, middle, and tail parts (Fig. 1). The tail parts of their names are simply ‘-mab,’ an acronym derived from their class of monoclonal antibodies. On the other hand, rozrolimupab, a mixture of recombinant human monoclonal Rhesus D antibodies, has a suffix of ‘-pab’ to indicate that it is a polyclonal antibody.
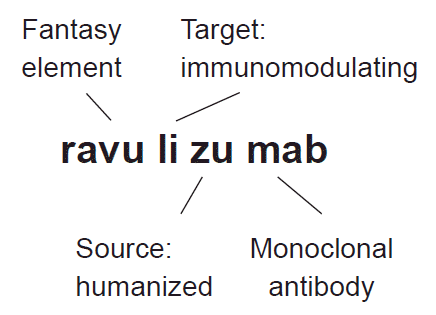 | Fig. 1.Example name of a monoclonal antibody. Target substem: ‘-li(m)-’ for immunomodulating, ‘-tu-’ for tumor target, ‘-ne-’ for neural target, ‘-ci-’ for cardiovascular target; source substem: ‘-ximab’ for chimeric monoclonal antibody, ‘-zumab’ for humanized monoclonal antibody, ‘-umab’ for fully humanized antibody, ‘-mab’ without source designation for newer monoclonal antibodies.
|
The middle part denotes the target and the source of the antibody. Rituximab, a chimeric antibody of murine Fab against CD20 and human Fc, was originally developed to treat lymphoma,8 and so it carries a substem of tumor-targeting (‘-tu-’) agent along with the tail for a chimeric monoclonal antibody (‘-ximab’). Ocrelizumab (Ocrevus®; Genentech, South San Francisco, CA, USA) is a humanized monoclonal antibody against the same CD20. However, its primary target was autoimmune disease, and so a substem for immunomodulating (‘-li-’) agents was added in front of the tail for a humanized antibody (‘-zumab’). Ofatumumab (Arzerra®; Novartis, Basel, Switzerland), another monoclonal antibody against CD20, is fully humanized, as reflected by the tail of its name (‘-umab’). Again, its primary target was chronic lymphocytic leukemia, although it was later approved for multiple sclerosis by the FDA. Likewise, infliximab (Remicade®; Janssen, Beerse, Belgium) and adalimumab (Humira®; AbbVie, North Chicago, IL, USA) are monoclonal antibodies against tumor necrosis factor alpha. As immunomodulators they share the infix of ‘-li(m)-,’ while they have tails denoting chimeric (‘-ximab’) and human (‘-umab’) antibodies,9 respectively. However, since most of the newer antibodies in current pipelines are humanized, the WHO decided to drop the source subsystem in 2017. Therefore, gosuranemab, a humanized anti-tau monoclonal antibody currently being tested in clinical trials, does not include a substem to denote its source. Other useful infixes include ‘-ne-’ for neural targets, as in galcanezumab (Emgality®; Eli Lilly, Indianapolis, IN, USA),10 and ‘-ci-’ for cardiovascular targets, as in abciximab (Reopro®; Eli Lilly, Indianapolis, IN, USA).
Go to :
RNA THERAPEUTICS11
RNA therapeutics are an emerging group of novel drugs. They are sequences of nucleotides or their analogues, designed to bind to a specific gene target. The naming system differentiates two underlying mechanisms of action: antisense oligonucleotides and RNA interference.
Antisense oligonucleotide drugs are a reverse-strand fragment of its targets. They can induce exon skipping, exon inclusion, or degradation by complementarily binding to the pre-mRNA in the nucleus. The common suffix for the antisense oligonucleotide products is ‘-rsen,’ reminiscent of their anti (reverse)-sense property; examples include eteplirsen (Exondys 51®; Sarepta, Cambridge, MA, USA)12 and viltolarsen (Viltepso®; Nippon Shinyaku, Kyoto, Japan)13 for Duchenne muscular dystrophy. The suffix of ‘-dirsen’ specifically indicates that the drug works for muscular dystrophy as in golodirsen (Vyondys 53®; Sarepta, Cambridge, MA, USA)14 and renadirsen. The suffix of ‘-nersen’ signifies that the drug is indicated for a neurological disorder, as in nusinersen (Spinraza®; Biogen, Cambridge, MA, USA) for spinal muscular atrophy.15
Antisense drugs adopt modified nucleotides to resist natural degradation by endogenous nucleases. Nusinersen has a 2’-O-2-methoxyethyl group in place of the 2’-hydroxy group of the RNA. Nonbridging oxygen in its phosphate backbone is replaced with a sulfur, to make a phosphorothioate link. In eteplirsen and viltolarsen, the ribose is replaced by a morpholino ring that extends through a phosphorus-nitrogen bond. These are technically not nucleotides, but rather phosphorodiamidate morpholino oligomers. Collectively, these molecules are better termed antisense oligomers.
Both inotersen (Tegsedi®; Ionis, Carlsbad, CA, USA)16 and patisiran (Onpattro®; Alnylam, Cambridge, MA, USA)17 inhibit transthyretin (TTR) expression but in two different ways. Inotersen is an antisense gapmer that has 2’-O-methoxyethyl-modified RNA at both ends and unmodified DNA in the middle, and it undergoes RNase-mediated degradation when bound to TTR pre-mRNA. Patisiran is a double strand of modified RNA oligomers that is packaged in a lipid complex. It enters the machinery of RNA interference and follows the pathway of small interfering RNA (siRNA) to degrade mature mRNA of TTR in the cytosol. The suffix of ‘-siran,’ an anagram of siRNA, is reserved for drugs that induce RNA interference.
Go to :
GENE AND CELL THERAPY18
Gene therapy products are biological drugs composed of a transgene and a vector that carries the gene. Each component is assigned a word: the first corresponds to the gene and the second corresponds to the vector. A historic drug, alipogene tiparvovec (Glybera®; UniQure, Amsterdam, Netherlands), was the first gene therapy agent indicated for an inherited disorder. It was approved in Europe for the treatment of lipoprotein lipase deficiency, but was discontinued in 2017 due to lack of demand. However, it was followed by voretigene neparvovec-rzyl (Luxturna®; Spark therapeutics, Philadelphia, PA, USA), which carries the gene encoding the enzyme retinoid isomerohydrolase (RPE65) and is indicated for the treatment of Leber congenital amaurosis; many gene therapy products are in the clinical development pipeline.
Onasemnogene abeparvovec-xioi (Zolgensma®; Novartis, Basel, Switzerland) is the first commercialized gene-therapy product for a neurological disorder (Fig. 2).19 The stem ‘-gene’ is common to all gene therapy drugs. The infix ‘-semno-’ derives from the gene symbol of survival of motor neuron (SMN), which it delivers, while the prefix ‘ona-’ is a fantasy word possibly derived from ‘one.’ The stem ‘vec’ of the second word denotes that the vector is a nonreplicating virus. Combined with the infix of ‘-parvo-,’ it indicates that the vector is an adeno-associated virus (AAV), which belongs to the virus family Parvoviridae. Again the prefix ‘abe-’ is a fantasy element given to identify a unique vector component.
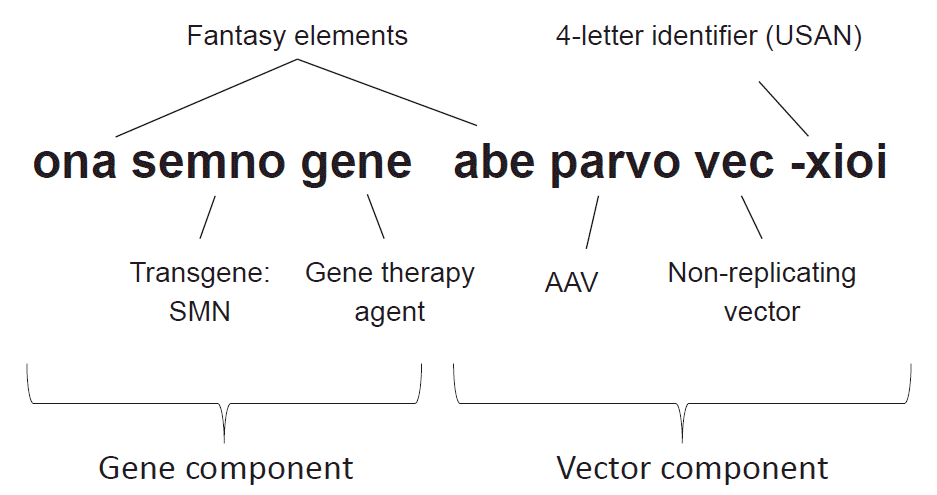 | Fig. 2.Example name of a gene-therapy agent. The gene component consists of fantasy element, transgene, and the suffix ‘-gene.’ The vector component also carries a fantasy element and announces its vector type. USAN, United States Accepted Name; SMN, survival of motor neuron; AAV, adeno-associated virus.
|
Lenzumestrocel (NeuroNata-R®; Corestem, Seongnam, Korea) is used in autologous bone-marrow mesenchymal stem-cell therapy.20 It is currently in the clinical trial for the treatment of amyotrophic lateral sclerosis. The suffix ‘-cel’ is common to all types of cell-therapy products, while the infix ‘-mestro-’ denotes that its origin is mesenchymal stromal cell.
Go to :
SMALL-MOLECULE THERAPIES21
Small-molecule drugs are old players that are expanding into new territories. Tyrosine-kinase inhibitors have a suffix of ‘-tinib,’ while ‘-nib’ is also used for other types of inhibitors. Protein kinases induce functional changes of their substrate protein by transferring a phosphate group. Inhibition of a specific protein kinase and its signaling pathway is a rapidly advancing type of cancer therapy.
Selumetinib (Koselugo®; AstraZeneca, Cambridge, UK) blocks the enzyme mitogen-activated protein kinase and is approved for the treatment of inoperable plexiform neurofibroma of neurofibromatosis type 1.22 Masitinib is another tyrosine kinase inhibitor, which modulates the activity of mast cells and macrophages; it is currently in clinical trials for the treatment of various conditions, including amyotrophic lateral sclerosis, multiple sclerosis, and Alzheimer’s disease. Other new small-molecule drugs are listed in Table 1.
Table 1.
Suffixes for new small molecules drugs
![]()
Go to :
CONCLUSION
Nomenclature of medicine is constantly being updated in pace with the development of new therapeutics. The system aims to provide universal and unequivocal names, while maintaining similarity between drugs with common properties. Having a comprehensive understanding of the nomenclature of new therapeutics is an indispensable tool for medical professionals.
Go to :
 XML Download
XML Download