

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Inflammatory myopathy is a chronic inflammatory muscle disease caused by immune-mediated muscle injury and consists of dermatomyositis (DM), polymyositis (PM), inclusion body myositis and immune-mediated necrotizing myopathy (IMNM). Patients with DM or PM present with progressive proximal muscle weakness over weeks to months and show good responses to immunosuppressive therapy [1]. DM is associated with distinct skin manifestations such as Gottron’s papules or a heliotrope eruption, which are useful in distinguishing between inflammatory myopathies. Additionally, other muscle diseases such as muscular dystrophy, metabolic, endocrine or drug-induced myopathies may mimic inflammatory myopathy, but none of these disorders are associated with the skin lesions characteristic of DM [2].
Limb-girdle muscular dystrophy 2B (LGMD2B), a subtype of dysferlinopathy, is characterized by predominantly proximal muscle weakness and markedly high creatine kinase (CK) levels [3]. LGMD2B, caused by the dysferlin gene (DYSF) mutation, usually occurs in childhood or early adolescence [4]. It may present as similar as inflammatory myopathy in terms of clinical, laboratory, and electromyography (EMG) features. Histologically, inflammatory cell infiltrations in myofibers could be present in LGMD2B [5]. Therefore, a late-onset presentation of LGMD2B can at times be mistaken for inflammatory myopathy.
Herein we report a case of a 48-year-old male with progressive proximal muscle weakness, papulosquamous lesions in the knuckles, elevated CK levels, and myopathic changes on the EMG. The initial impression of his diagnosis was DM, yet the final diagnosis turned out to be LGMD2B with psoriasis.
Go to : 
CASE REPORT
A 48-year-old male presented with proximal muscle weakness in his lower extremities followed by skin rashes on the hands. Muscle weakness had slowly progressed over the past eight months. He had difficulty in sitting up and could only move around using a wheelchair or a walker. He denied myalgia, arthralgia, or dysphagia. He was diagnosed with type 2 diabetes mellitus, hypertension, and dyslipidemia during the past three years. He had a second degree burn over 70% of his face ten years ago. He was a 15-pack-year smoker with no occupation and drank alcohol once a week. He was taking aspirin, atorvastatin, perindopril and nateglinide. His family history of medical illness was unremarkable.
On physical examination, his vital signs were stable, and the body mass index was 19.8 kg/m2 (height 165 cm, body weight 54 kg). Proximal muscle strength in both lower extremities was grade 3 out of 5 based on the Medical Research Council scale. The strength in his distal lower extremities and the proximal and distal upper extremities was grade 4+. Calf muscle atrophy was not observed. Erythematous papulosquamous skin lesions with scales were present on the dorsal surface of his metacarpophalangeal joints, fingers, and elbows. Physical findings in other systems and organs were unremarkable. At his first visit, CK level was 4,512 IU/L (normal 45∼163), aldolase was 18.2 U/L (0∼7.6), and lactate dehydrogenase was 432 IU/L (100∼225). Aspartate transaminase and alanine transaminase levels increased to 75 IU/L (1∼40) and 77 IU/L (1∼40), respectively. His complete blood count was as follows: white blood cell count 6,220 /μL, hemoglobin 14.7 g/dL, and platelets 267× 103 /μL. The erythrocyte sedimentation rate and C-reactive protein levels were 8 mm/hr (0∼9) and 1.57 mg/dL (0∼0.5), respectively. Anti-nuclear antibody, rheumatoid factor, and anti-Jo-1 antibody were not detected. EMG revealed small-amplitude, short-duration and polyphasic motor-unit potentials without increased spontaneous activity in the deltoid, biceps brachii, and pronator teres, and high amplitude and long duration motor-unit potentials in the vastus lateralis, tibialis anterior, and gastrocnemius muscles. Nerve conduction studies were normal. Results of the electrocardiogram and chest radiograph were unremarkable. There was no evidence of malignancy on further evaluation. For economic reasons, muscle imaging or biopsy was not initially performed.
With the suspicion of DM (symmetric proximal muscle weakness, scaly patchy redness over knuckles, elevated muscle enzyme levels, and EMG abnormalities), 20 mg/day of prednisolone and 300 mg/day of hydroxychloroquine were given. However, muscle weakness did not respond to the initial combination of medications within 6 months. Erythematous papules and plaques with silvery scales on his elbows, hands, and legs were confirmed as psoriasis by a Dermatologist; topical ointment and 12.5 mg/week of methotrexate were prescribed.
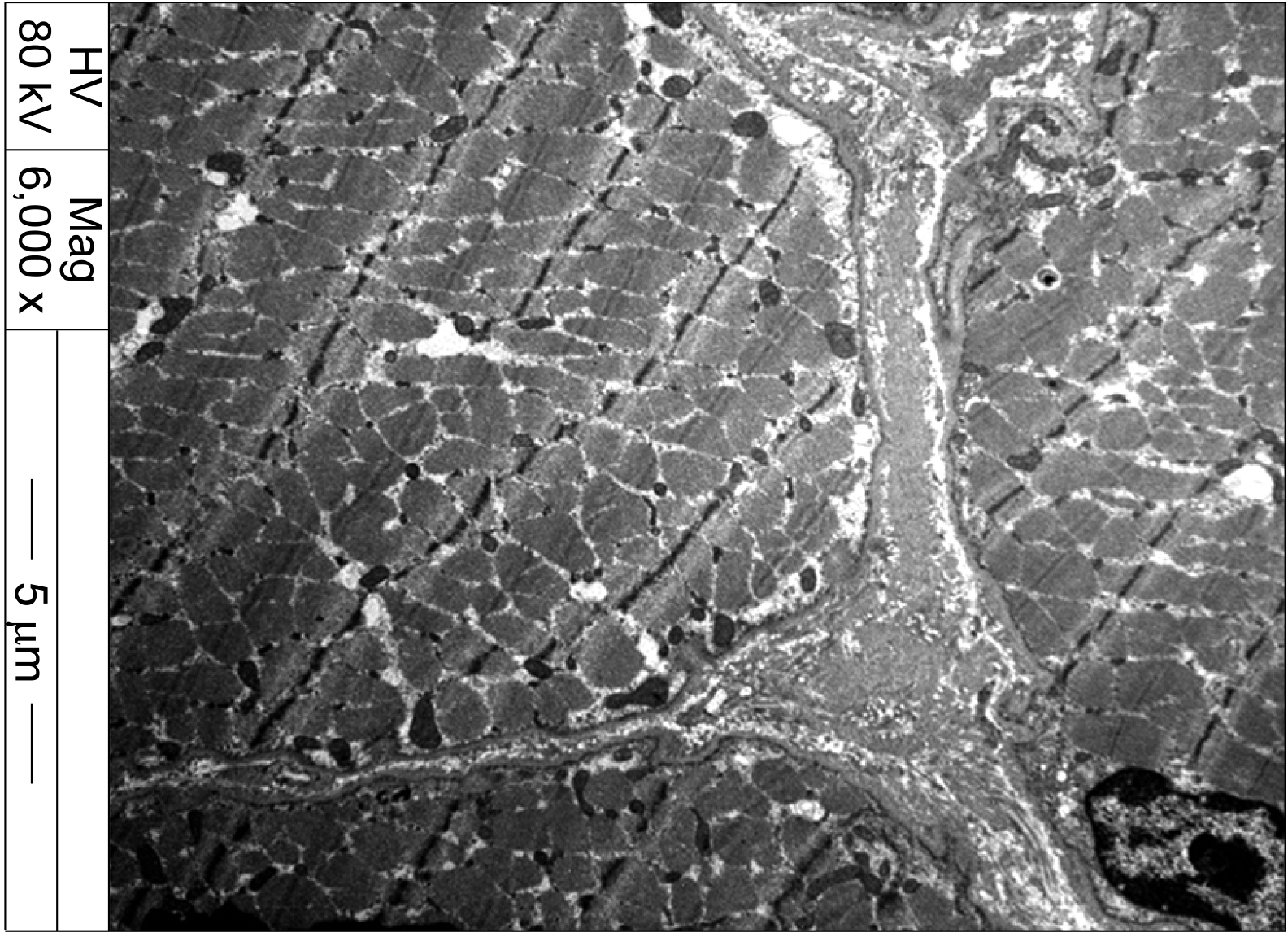
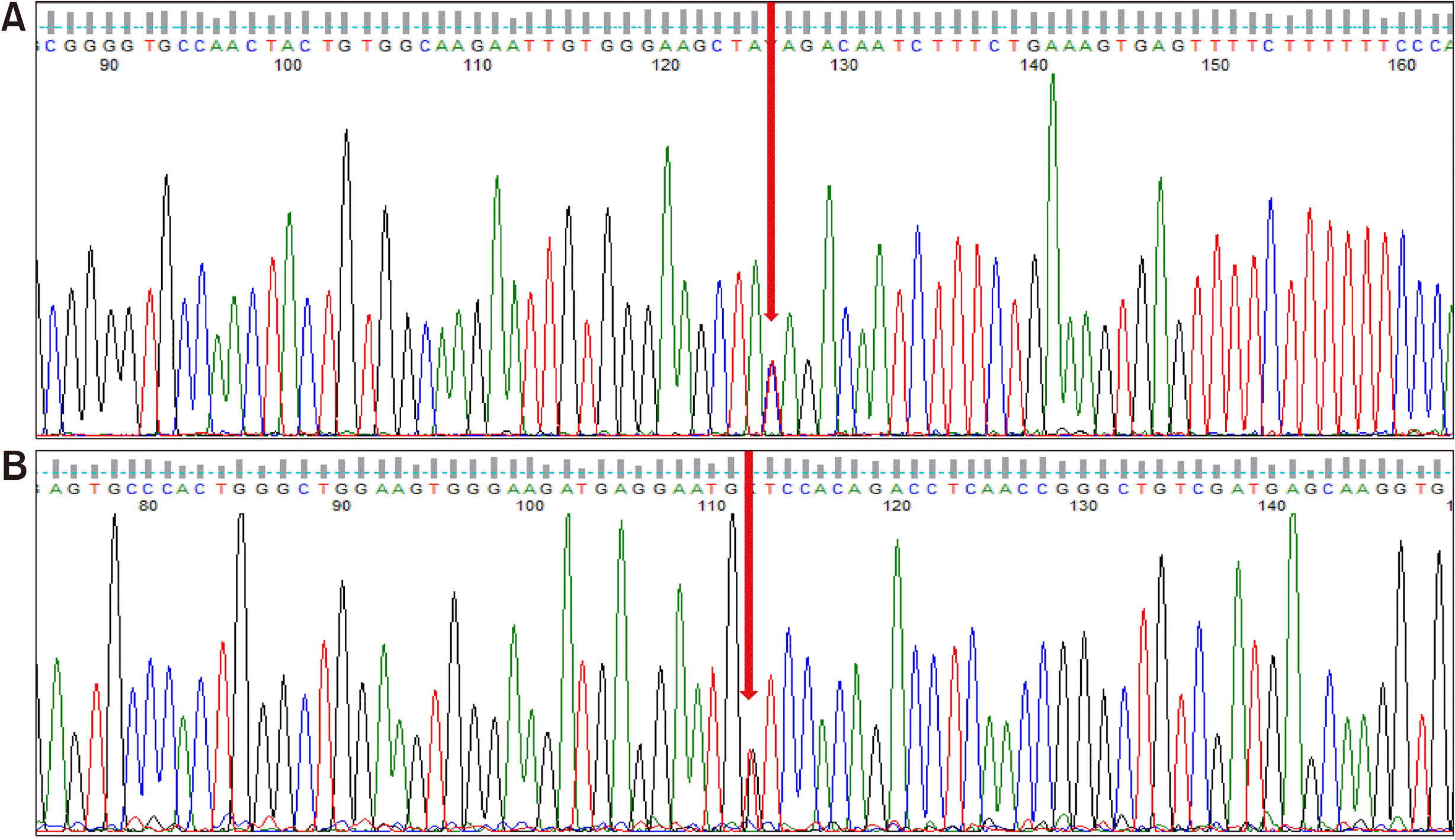
Despite the immunosuppressive treatment, muscle weakness continued to worsen and CK levels fluctuated throughout the following 12 months (Figure 1). Eventually, a muscle biopsy of the vastus lateralis was performed. The specimen showed muscle cell degeneration, as well as regeneration and atrophy. Fatty replacement of muscle tissue was severe. There were no muscle cell necrosis or inflammatory cell infiltrates in endomysial and perivascular regions. Neural cell adhesion molecule (CD56) and dystrophin were present, yet dysferlin was absent on immunostaining (Figure 2). Electron microscopy did not show any tubuloreticular inclusion (Figure 3). Furthermore, a molecular analysis with Next Generation Sequencing disclosed heterozygous mutations in the DYSF gene; c.2548C>T (p.Gln850*) and c.3051G>T (p.Trp1017Cys) [6]. Sanger sequencing confirmed that the mutations segregated with the disease phenotype (Figure 4). A final diagnosis of LGMD2B was made, and all immuno-suppressive agents were tapered off. The patient remained stable on physiotherapy.
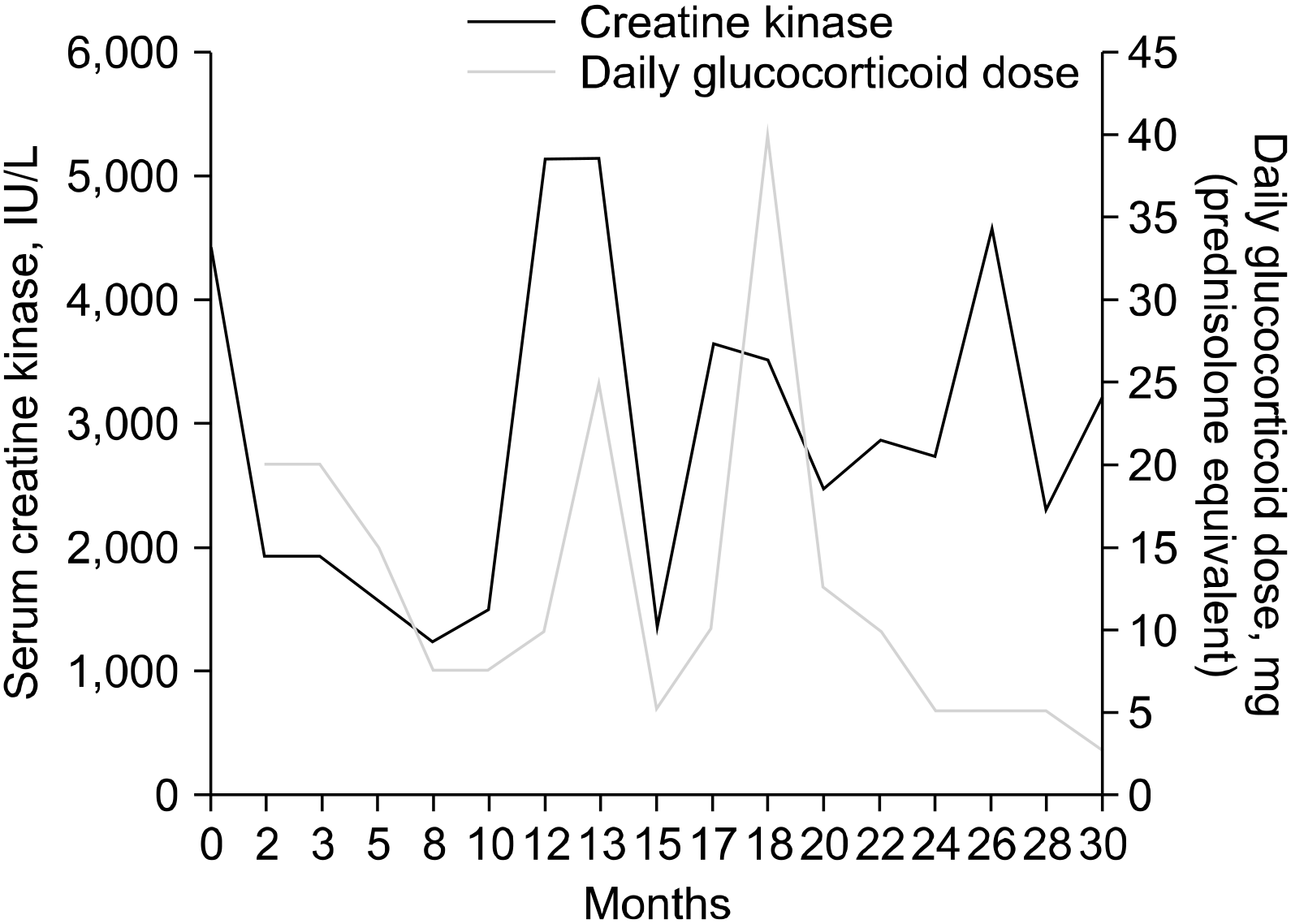 | Figure 1Changes in serum creatine kinase levels and daily doses of glucocorticoid during the follow-up period.
|
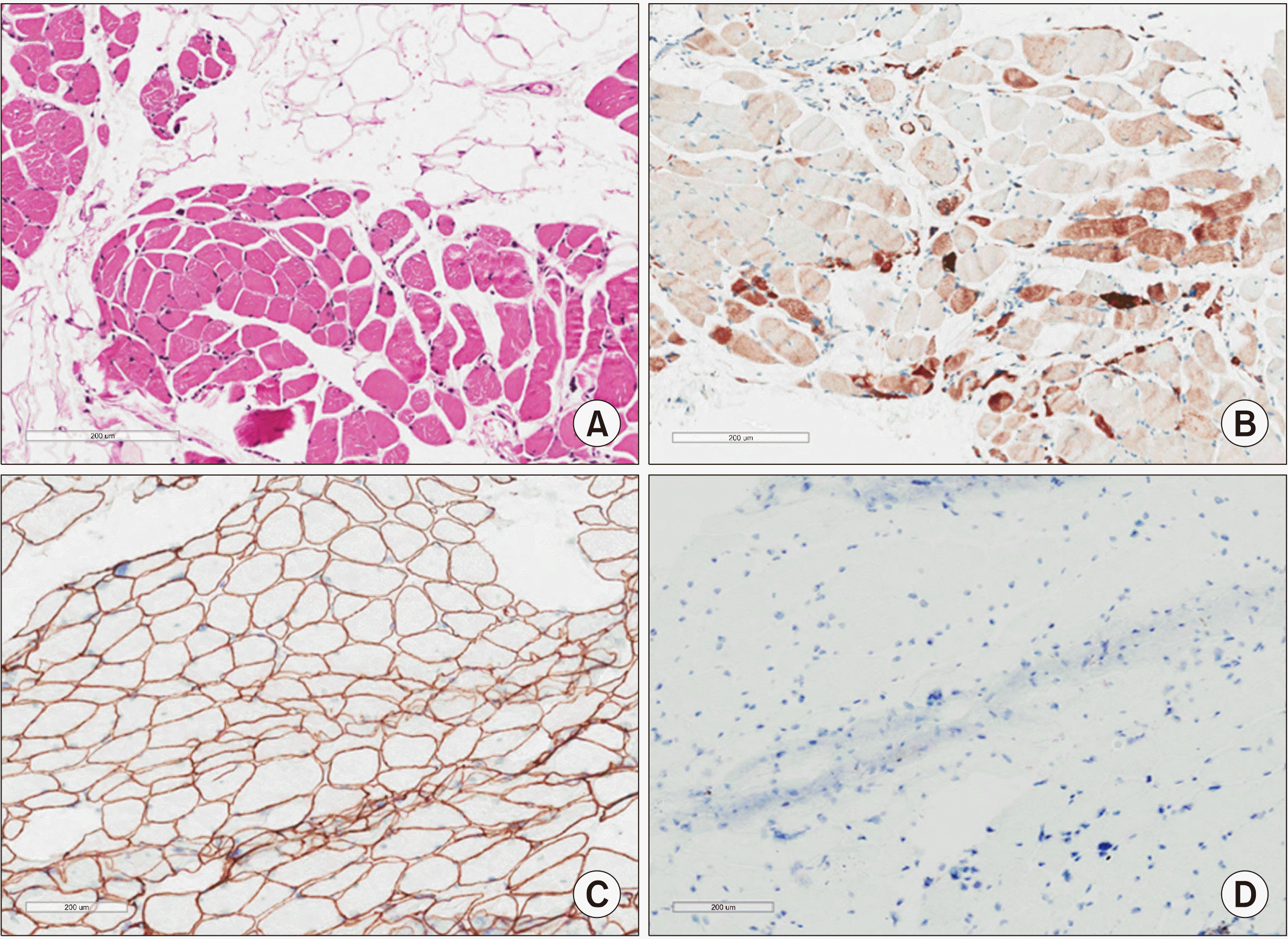 | Figure 2H&E stain (A) and immunohistochemistry (B∼D) of the muscle biopsy. (A) Myofibers show moderate size variation with degenerating cells with endomysial fibrofatty change (H&E, ×200). (B) Many degenerated myofibers show variation of size and positivity for CD56 (CD56 immunohistochemistry, ×100). Dystrophin (C) was robustly positive (Dystrophin 1 immunohistochemistry, ×200), yet dysferlin (D) was totally absent in the sarcolemmal membrane (Dysferlin immunohisto-chemistry, ×200).
|
The study was approved by the Institutional Review Board of Seoul Metropolitan Government-Seoul National University Boramae Medical Center (IRB No. 16-2017- 61). Informed consent was obtained from the patient.
Go to :
DISCUSSION
Our case presents a 48-year-old male with progressive proximal muscle weakness in the lower limbs, distinctive skin rashes, high CK levels, and a myopathic pattern in the EMG. He was initially diagnosed with DM, but showed poor treatment response to immunosuppressive agents. After dysferlin immunostaining of the muscle tissue and genetic analysis, a final diagnosis of LGMD2B was made.
Dysferlin, a protein encoded by the DYSF gene located on chromosome 2p13, plays a role in vesicle docking and fusion with the plasma membrane to repair sarcolemma disruption. Thus, mutations in the DYSF gene are responsible for a defective repair mechanism in the sarcolemma leading to muscular dystrophy: a LGMD2B affecting the proximal lower limbs at the onset, and a Miyoshi myopathy (MM) affecting posterior distal compartment of the legs [7]. Both LGMD2B and MM have similar features, however, muscle weakness with pelvic girdle distribution distinguishes between LGMD2B and MM. Interestingly, the type of DYSF gene mutation does not correlate with phenotypic features of dysferlinopathy and shows clinical heterogeneity even with the same mutation [8]. Patients with dysferlinopathy, especially LGMD2B, have symmetric proximal weakness, high CK levels and small polyphasic motor-unit potentials in the EMG, which may be confused with inflammatory myopathy. Nguyen et al. [4] reported that approximately 25% of patients with dysferlinopathy are initially misdiagnosed with inflammatory myopathy. Nevertheless, the two entities display several different clinical features. First, patients with dysferlinopathy commonly demonstrate inability to stand or walk on toes due to distal muscle involvement and atrophy of the gastrocnemius [9]. Second, the disease onset of dysferlinopathy usually occurs during early adulthood (typically 15 to 35 years), and patients may have a pre-clinical period with relatively high CK levels ranging from 1,000 to 40,000 IU/L [3,10]. Third, treatment response to glucocorticoid and/or other immunosuppressive agents is far from adequate. The mainstay management of dysferlinopathy is exercise to maintain muscle strength [11]. Table 1 compares the characteristics of dysferlinopathy with those of inflammatory myopathies.
Table 1
Differential diagnoses of dysferlinopathy
![]()
The precise distinction between dysferlinopathy and inflammatory myopathy is based on muscle biopsy and dysferlin immunostaining, which shows a significant reduction or absence of dysferlin protein in the sarcolemma in dysferlinopathy [12]. Interestingly, inflammatory cell infiltrates in muscles are often present in dysferlinopathy [13]. Inflammatory cell types in dysferlinopathy shows high CD4+/CD8+ T cell ratios predominantly in the perivascular region similar to DM. But unlike inflammatory myopathy, major histocompatibility complex class I expression is low and sarcolemmal membrane attack complex is deposited on non-necrotic fibers [5]. In our case, IMNM should also be considered since the patient was on statin therapy, but no muscle cell necrosis or macrophage infiltration was found in the biopsy specimen [14].
Our patient demonstrated several unusual features of dysferlinopathy that made the diagnosis difficult. First, age of onset was later in life compared with that presented in previous studies. Park et al. [15] reported that the age of onset varied from 9 to 36 years in Korean patients with dysferlinopathy. A Japanese study showed a wider range of age; 14 to 58 years [16]. Late-onset LGMD2B is associated with the homozygous mutation of the c.2997G>T (p.Trp999Cys) allele in Japanese [16]. Two of 23 Korean patients with dysferlinopathy showed a heterozygous mutation in the c.2997G>T allele; one was a 41-year-old male with Miyoshi myopathy and the other was a 27-year-old female with LGMD2B [6]. The mutations in our patient and its association with a late-onset phenotype need to be further investigated. Second, psoriasis sporadically appeared in multiple regions in our patient along with muscle weakness. There is no report on the association between dysferlinopathy with psoriasis to date. It is sometimes challenging to distinguish between psoriasis and Gottron’s papules seen in DM as the two often develop at similar anatomical sites.
Go to :
SUMMARY
In summary, due to the clinical heterogeneity of dysferlinopathy and its overlapping features with inflammatory myopathy, it could sometimes be difficult to discern the two entities. Dysferlinopathy should be considered in patients with myopathy who have markedly high CK levels, calf muscle atrophy, and poor response to immuno-suppressive therapy. Muscle dysferlin immunostaining plus genetic analysis would be essential for its diagnosis.
Go to :
 XML Download
XML Download