

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Langerhans cell histiocytosis (LCH) is a myeloproliferative disorder characterized by inflammatory lesions with the accumulation of CD1a+/CD207+ histiocytes, leading to the destruction of affected tissues [1, 2]. While LCH invades bones and skin commonly, it can involve any organ, including the liver, spleen, lungs, lymph nodes, central nervous system, and hematopoietic system. The clinical course varies greatly from a single bone lesion to a fatal multi-organ disease [3]. Although the precise origin of the pathological histiocytes in LCH is unclear, the current hypothesis is that these pathological cells are more likely to arise from dysregulated differentiation or the recruitment of bone marrow-derived precursor cells or yolk sac progenitors than from epidermal Langerhans cells [4, 5]. Recent advances in genomic sequencing have highlighted the activation of the mitogen-activated protein kinase (MAPK) pathway at critical stages in myeloid differentiation as an essential driver of LCH pathology [4]. In this review, we overview the recently growing understanding of LCH pathogenesis and a novel therapeutic option to optimize and personalize therapy through improved risk stratification, targeted therapy, and the assessment of therapy responses based on specific molecular features.
Pathological considerations of LCH
Diagnosis and clinical overview of LCH: Histiocytic disorders are diseases associated with cells of the mononuclear phagocyte system, which include hematopoietic cells with mononuclear morphology and phagocytic activity. LCH is the most common histiocytic disorder, and its clinical presentation varies from limited, indolent, and self-regressive to life-threatening disseminated disease. The diagnosis of LCH is based on clinical and radiological findings in combination with histopathological analysis, identifying the infiltration of tissue by histiocytes using the ultrastructural or immunophenotypic characteristics of Langerhans cells, which include the surface expression of CD207 (Langerin) and CD1a, as well as the detection of Birbeck granules using electron microscopy [6]. In routine clinical practice, the identification of Birbeck granules has recently been replaced with positive immunohistochemical staining for CD1a and CD207.
LCH as an immunoreactive disorder versus as a neoplastic disorder: In LCH lesions, pathological CD1a+CD207+ histiocytes are present in a small proportion, from less than 1% to more than 70% (median 8%), and are mixed with a variety of inflammatory cells, including eosinophils, macrophages, multinucleated giant cells, and lymphocytes (enriched with regulatory T cells) [7, 8]. In addition, these pathological histiocytes have a benign morphology and low mitotic activity. In this regard, LCH is considered an immune disorder in which normal cells proliferate in response to environmental stimuli.
The findings of Badalian-Very et al. [9], in 2010, showed that 57% of patients with LCH had recurrent BRAF-V600E mutations, and subsequent studies have identified recurrent somatic mutations in pathological LCH cells, revealing the neoplastic nature of LCH and leading to the definition of LCH as an inflammatory myeloid neoplasm [1].
Misguided myeloid differentiation model of LCH pathogenesis: The identification of the BRAF-V600E mutation provided a molecular tag for lineage tracking to test the hypothesis that LCH originated from early myeloid progenitors rather than epidermal Langerhans cells. An analysis of peripheral blood mononuclear cells (PBMCs) identified the presence of a small (<1%) but consistent proportion of BRAF-V600E mutations in myeloid cells (CD11c+ myeloid dendritic cell precursors and CD14+ monocytes) in patients with high-risk LCH. Similarly, in bone marrow samples, BRAF-V600E was identified in CD34+ hematopoietic stem cells from many patients with high-risk LCH, of which up to 50% were reported to have normal histology. In contrast, BRAF-V600E was generally absent in PBMCs from patients with single lesion LCH and rare in the PBMCs of patients with multifocal low-risk LCH [4, 7, 10]. These observations support the “misguided myeloid differentiation model” of LCH, where the extent of disease is defined by the differentiation stage of the cell in which the activating somatic MAPK mutation occurs (Fig. 1) [4]. According to this model, the high-risk multisystem LCH is caused by somatic activating mutations in the MAPK gene of hematopoietic stem/progenitor cells from bone marrow; the low-risk multisystem LCH is caused by somatic MAPK mutations of committed dendritic cell (DC) precursor cells in the blood, and the low-risk single-system LCH is caused by somatic MAPK mutations of more differentiated DC precursors from blood residing in a single lesion [4, 7, 10].
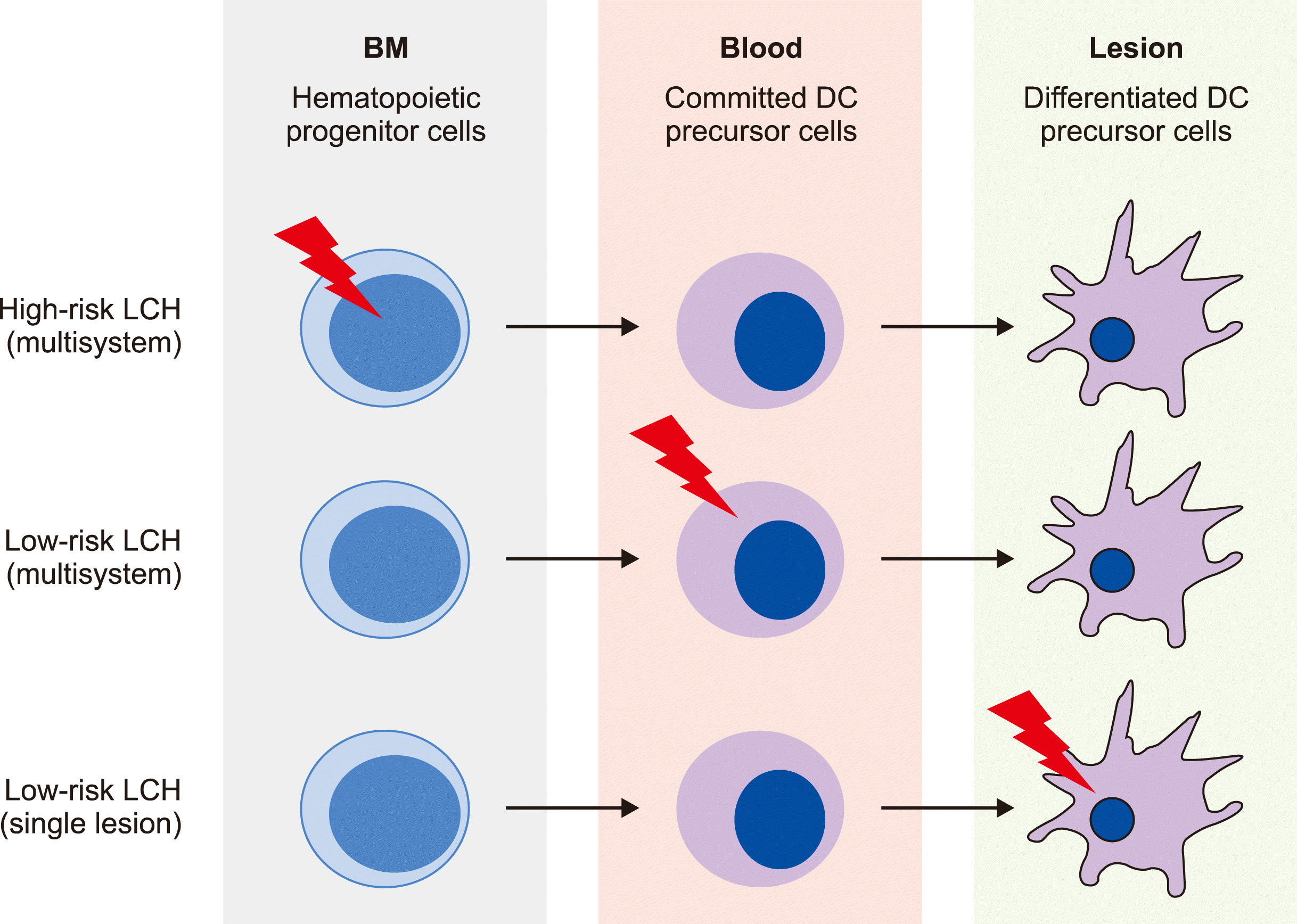 | Fig. 1Misguided myeloid differ-entiation model for LCH ontogeny according to different risk groups. According to this model, the extent of disease is defined by the differ-entiation stage of the cell in which the activating somatic MAPK mutation occurs.
Abbreviations: DC, dendritic cell; LCH, Langerhans cell histiocytosis.
|
Recent progress in molecular pathogenesis of LCH
Activation of MAPK pathway in LCH: The improvement of sequencing techniques that amplify the sequencing depth has enabled simultaneous studies of multiple target genomic regions per sample, which has led to the identification of the recurrent oncogenic somatic mutations in LCH that activate the MAPK pathway. The MAPK pathway comprising RAS/RAF/MEK/ERK, which regulates cell differentiation, proliferation, and apoptosis, plays a vital role in myeloid cell differentiation and maturation (Fig. 2). Previous reports have shown evidence of activation of the MAPK pathway in all the LCH samples [9, 11]. Mutations in the MAPK pathway proteins are associated with the differentiation of DC progenitors and inhibit the maturation of DCs. This role of activation of the MAPK pathway in myeloid cell differentiation and maturation may have implications regarding the origin and functional alterations of histiocytes in LCH [4].
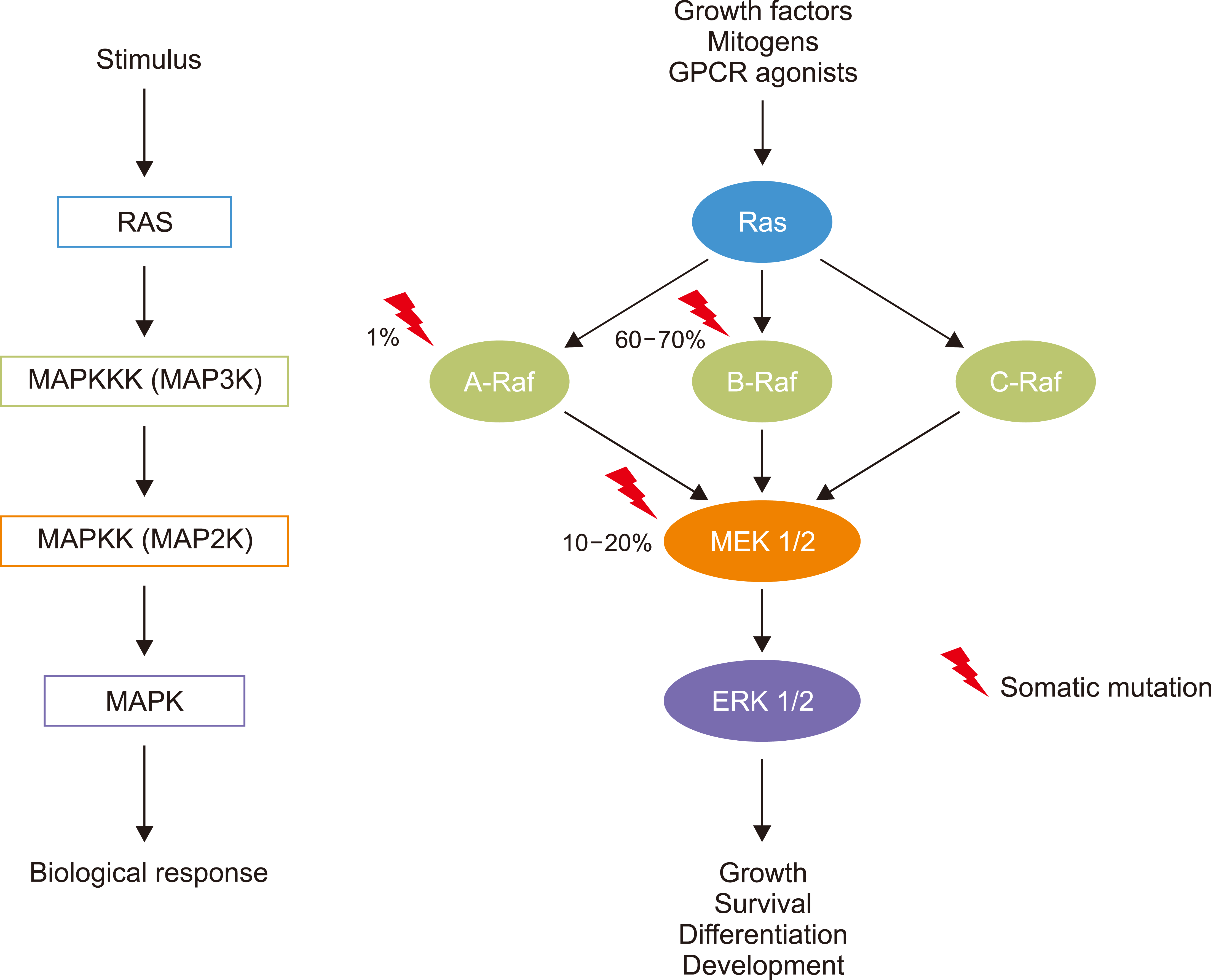 | Fig. 2MAPK pathway mutations in LCH. Activating MAPK pathway mutations in LCH drive pathologic ERK activation.
Abbreviations: A-Raf, rapidly accelerated fibrosarcoma isoform A; B-Raf, rapidly accelerated fibrosarcoma isoform B; C-Raf, rapidly accelerated fibrosarcoma isoform C; ERK, extracellular signal-regulated kinase; GPCR, G-protein-coupled receptor; LCH, Langerhans cell histiocytosis; MAPK, mitogen-activated protein kinase; MEK, mitogen-activated protein kinase kinase; Ras, rat sarcoma.
|
Somatic mutations associated with LCH: Badalian-Very et al. [9] identified the BRAF-V600E mutation in pathological LCH cells and subsequent studies explored the genomic landscape of childhood LCH using targeted and whole-exome next-generation sequencing (NGS) approaches. The heterozygous BRAF-V600E mutation is a predominant genomic alteration in LCH, estimated in 38–64% of patients with LCH [7, 9, 12, 13]. Recurrent mutations in exons 2 and 3 of the MAP2K1 gene, which encodes MEK1, a downstream signaling protein of BRAF in the MAPK pathway, were reported in 15–20% of LCH cases [14, 15]. In addition, small in-frame BRAF deletions or insertions localized to exon 12, which encodes the small N-terminal lobe of the BRAF kinase domain, have been reported in 5–10% of LCH cases [14, 16]. In addition, rare cases of mutations of other genes encoding MAPK pathway proteins have been reported, such as FAM73A-BRAF fusion [14], MAP3K1 mutation [15], and ARAF mutation [17]. A somatic oncogenic PIK3CA mutation associated with the PIK3-AKT pathway has been reported in a pediatric LCH case and in an adult non-LCH disease (Erdheim-Chester disease) [18, 19]. No genetic alterations were found in approximately 20% of the patients with LCH. In these cases, copy number changes or epigenetic alterations may be additional mechanisms for MAPK pathway activation [1].
Targeted therapy in LCH
BRAF kinase inhibitors: vemurafenib and dabrafenib: Identification of BRAF and MAP2K1 mutations in LCH led to targeted therapies acting on the MAPK pathway. First-generation BRAF inhibitors, vemurafenib and dabrafenib, for the treatment of high-risk or refractory LCH, have been studied. The efficacy of BRAF inhibitors in the treatment of LCH was first reported in the off-label clinical use of vemurafenib in three patients with multisystem and refractory ECD carrying the BRAF-V600E mutation, two of whom also had combined LCH, and the other being a young infant with refractory, multisystem LCH. In these reports, patients receiving vemurafenib showed a dramatic response and good short-term tolerability [20, 21]. A recent international study of vemurafenib in 54 pediatric patients with refractory multisystem LCH showed a 100% overall response rate (complete response in 70% and partial response in 30%) after at least eight weeks of vemurafenib (20 mg/kg/day) monotherapy. However, 80% of the patients experienced LCH reactivation after the discontinuation of vemurafenib. The re-introduction of vemurafenib was effective in 75% of the reactivated patients [22]. Previous studies on vemurafenib have found that while patients achieved excellent response rates, frequent recurrences were observed after the discontinuation of vemurafenib [23, 24]. Dabrafenib is another treatment option for high-risk and refractory LCH. Dabrafenib is a selective BRAF kinase inhibitor that has been reported to have fewer adverse effects [25]. In a recently reported Chinese pediatric study, with 20 enrolled patients with refractory LCH that were treated with dabrafenib monotherapy, the overall objective response rate was 65%, and the overall disease control rate was 75% [26].
Drawbacks of BRAF kinase inhibitors and future perspectives of target therapy: Although BRAF-V600E targeted therapy is an important advance, especially for refractory or high-risk LCH, the use of BRAF kinase inhibitors has several drawbacks that need to be overcome. BRAF inhibitors were initially used in the treatment of metastatic melanoma and other adult BRAF-mutated cancers. According to the adult experience of BRAF inhibitors, BRAF inhibitors were ineffective for many other BRAF mutations, such as BRAF fusion with a partner gene, insertion/deletion of exon 12, and ineffective in activating mutations of genes that encode proteins acting upstream of RAF (such as RAS) [27]. This ineffectiveness can trigger a paradoxical transactivation mechanism, which is responsible for serious cutaneous adverse effects, including keratoacanthomas and squamous-cell carcinomas [28]. In pediatric cases, rapid LCH reactivation has been observed after the discontinuation of BRAF inhibitors, and it has been found to be ineffective in sclerosing cholangitis and advanced neurodegenerative disease [1, 22, 29]. In this regard, prospective trials should be performed to determine an optimal duration of use and potential combinations with other targeted or cytotoxic therapies. An approach combining a BRAF inhibitor with a MEK inhibitor that is used in adult malignancies can be effective in LCH. There is currently an ongoing international clinical trial to evaluate the efficacy of a combination of dabrafenib and the MEK inhibitor, trametinib, for adults and children with refractory or relapsed LCH (MEK inhibitor±BRAF inhibitor, NCT02124772).
Clinical considerations according to molecular pathogenesis
BRAF-V600E mutation as a risk factor of LCH: Several previous studies have shown that the BRAF-V600E mutation is associated with the severity of LCH and with treatment failure. Berres et al [7]. reported that patients with a BRAF-V600E mutation had an approximately two-fold risk of reactivation. Héritier et al. [30] found that patients with BRAF-V600E had more severe disease, tended to resist standard therapy, had a higher reactivation rate, and more permanent, long-term sequelae after treatment. In this regard, the French group recommended that a molecular analysis be performed at least once for the following presentations at the initial biopsy: 1) LCH with risk organ involvement; 2) LCH diagnosis before two years of age; 3) LCH with pituitary involvement or skull base or orbit bone lesions; 4) resistance to first-line therapy; and 5) severe complications and sequelae [1].
Circulating cell-free BRAF-V600E as a biomarker in LCH: Héritier et al. [30] quantified the BRAF-V600E allele in the circulating cell-free DNA from plasma samples from pediatric patients with LCH. This study reported a higher BRAF ccf load in patients with risk organ involvement and a sequential decrease in the BRAF ccf load after treatment [30]. Recent studies of BRAF inhibitors in patients with LCH included follow-up tests of BRAF ccf load and showed that BRAF ccf load reduced rapidly after BRAF inhibitor treatment [22, 26]. These results indicated that the BRAF ccf load was associated with high-risk features and was responsive to chemotherapy. In this regard, the ccf BRAF load should be considered as a blood biomarker of LCH in future clinical trials.
Go to : 
CONCLUSION
LCH has long been an enigma with respect to its origin and pathogenesis. Recent advances in molecular genomic analysis techniques have revealed the pathogenic molecular mechanism of LCH. The understanding of the origin and biology of LCH has increased rapidly in the last decade, leading to the introduction of promising targeted therapies for high-risk patients. However, high-risk patients still have suboptimal outcomes, and targeted therapy for such patients cannot eradicate the malignant clone and provide long-lasting remission after discontinuation. Future research should focus on improving outcomes and reducing permanent sequelae in high-risk LCH patients based on the recent understanding of LCH.
Go to :
 XML Download
XML Download