

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Chronic myelomonocytic leukemia (CMML) is a type of clonal hematopoietic disease characterized by an increase in monocytes and dysplasia of myeloid precursor cells. It is classified as a myelodysplastic syndrome/myeloproliferative neoplasm (MDS/MPN) along with atypical chronic myeloid leukemia (CML), juvenile myelomonocytic leukemia, MDS/MPN with ring sideroblast and thrombocytosis, and unclassified MDS/MPN based on the World Health Organization (WHO) classification revised in 2016 [1]. CMML is common in old age, show a male predominance, and is a relatively rare disease. Although the exact incidence is unknown, it is estimated to be approximately 1 in 100,000 [2, 3]. CMML does not have a pathognomonic finding that is crucial for diagnosis, such as BCR-ABL1 rearrangement in CML. It is diagnosed by excluding other diseases accompanied by monocytosis and other types of MDS/MPN diseases. In addition, CMML is composed of several heterogeneous subgroups, represented by the dysplastic and proliferative types, and these various phenotypes differentially affect the treatment outcomes and disease prognosis.
The accumulated clinical information and laboratory data on CMML show the evolution in the diagnosis and treatment of the disease. In recent years, developments in DNA sequencing technology have enabled the identification of many genetic features of blood diseases. Therefore, in CMML, in addition to the existing morphologic findings, molecular signatures have started to be used for diagnosis and risk prediction [4-6]. The treatment of CMML is still mainly based on cytoreductive therapy, hypomethylating agents (HMAs), and allogeneic hematopoietic stem cell transplantation (HSCT), although various changes are expected with the development of several novel agents with therapeutic potential in the field of hematology [7-9].
This review summarizes the diagnostic criteria, prognostic scoring systems, and treatment options for CMML based on the results of several recently published studies and revised guidelines.
DIAGNOSTIC APPROACH AND RISK ASSESSMENT
Diagnostic criteria
For the diagnosis of CMML, the essential methods include peripheral blood tests, bone marrow aspiration and biopsy, chromosomal analysis, and genetic tests. Among them, the morphologic evaluation of peripheral blood and the bone marrow is the basis of CMML diagnosis. The hallmark of CMML is dysplastic change in myeloid-lineage cells in the bone marrow accompanied by a persistent and considerable increase in monocyte counts in peripheral blood.
The diagnostic criteria for CMML according to the 2016 WHO classification are described in Table 1 [1]. CMML is further classified into 3 subtypes according to the percentage of blasts in peripheral blood and the bone marrow. It is also divided into the dysplastic and proliferative types according to the white blood cell (WBC) count in peripheral blood. These 2 subtypes differ in clinical manifestations and complication patterns. The proliferative type is associated with constitutional symptoms and more often presents with organomegaly, whereas the dysplastic type has more severe symptoms due to cytopenia, including anemia and thrombocytopenia [10, 11]. It is known that mutations in genes involved in the regulation of rat sarcoma (RAS) signaling pathways contribute to conversion from the dysplastic type to the proliferative type [12].
To diagnose CMML, an approach to rule out other diseases accompanied by monocytosis is also required. To exclude CML with BCR-ABL1, a typical disease accompanied by an increase in WBCs including monocytes and the appearance of immature cells in peripheral blood, the BCR-ABL1 fusion gene should be identified. In addition, when monocytosis and significant eosinophilia are observed together, the rearrangement of PDGFRA and PDGFRB should be confirmed. Patnaik et al. [13] reported that among patients with features similar to CMML, 1 patient with t(5;12)(q31-q32;p13) and ETV6(TEL)-PDGFRB fusion oncogenes had eosinophilia. Meanwhile, in a patient diagnosed with CMML-2, there was no eosinophilia but an SDPR-PDGFRB rearrangement was found, and this patient showed a short and partial response to imatinib [14]. Patients harboring rearrangements associated with the PDGFR gene are distinguished from those with classical CMML by their excellent responses to tyrosine kinase inhibitors such as imatinib. Therefore, although these diseases are similar to CMML in morphologic aspects, they are currently classified into separate disease categories.
Some patients with cytopenia do not meet the diagnostic criteria of CMML but have relative monocytosis and dysplastic features of the bone marrow. They are younger and have a lower WBC count than those with classical CMML but share the characteristics of CMML, such as splenomegaly, anemia, thrombocytopenia, and a high frequency of TET2 and SRSF2 gene mutations [15, 16]. They are classified as having MDS or unclassified MDS/MPN according to the current WHO classification; however, the disease is converted to CMML at a high frequency during the follow-up period and can even progress to secondary acute myeloid leukemia (AML). Therefore, Valent et al. [17] classified this condition as oligomonocytic CMML, which is a precursor of CMML, and suggested diagnostic criteria for this disease category.
Immunophenotyping
The increase in monocytes is a necessary condition for CMML; however, increased monocytes can have various reasons. Therefore, until a patient with monocytosis is confirmed to have CMML, a differential diagnostic process that assumes various causes is required.
Conditions in which monocytes are increased in peripheral blood need to be distinguished from reactive monocytosis and increased monocyte counts caused by other reasons such as inflammatory processes or MPN. The classification of monocytes using multiparameter flow cytometry (MPFC) is useful for discriminating CMML. Human monocytes are classified into 3 subtypes according to the expression patterns of the surface markers CD16 and CD14 [18]. Classical monocytes are strongly positive for CD14 and negative for CD16 and account for the majority of monocytes in the blood of healthy humans. Intermediate monocytes are positive for both CD14 and CD16, whereas nonclassical monocytes are weakly positive for CD14 and positive for CD16. It is known that the proportion of classical monocytes is increased in CMML patients. Further, when the cutoff value of the percentage of classical monocytes in MPFC is 94%, both the sensitivity and specificity for the diagnosis of CMML are >90%. In addition, a <1.13% fraction of nonclassical monocytes is also useful to distinguish CMML from other diseases [19-21]. This pattern, revealed by MPFC findings, is independent of the CMML subtype, karyotype, and mutational factors. In addition, MPFC could be used for evaluating the treatment response because it has been found that CMML patients who achieve a treatment response to HMAs show normalization of monocyte count on MPFC after treatment [19]. However, there are some limitations to the practical application of this approach in clinical practice: false-negative results can occur in the presence of accompanying inflammatory conditions including autoimmune diseases and false-positive results can arise from other myeloid neoplasms such as atypical CML [22].
Cytogenetics
Abnormal chromosomal types are observed in approximately 25–40% of CMML patients [23-26]. The types of chromosomal abnormalities associated with CMML do not vary, although the most common types are trisomy 8, loss of Y (–Y), and monosomy 7. The association between chromosome type and CMML prognosis is well known. In most studies, the risk according to karyotype has been classified into 3 stages. Spanish researchers suggested a CMML-specific cytogenetic risk classification [24]. In this system, patients with a normal karyotype and –Y comprise the low-risk group, those with trisomy 8 and 7 chromosome abnormalities and complex karyotypes are considered the high-risk group, and those with all other chromosome types are classified into the intermediate-risk group. The median survival period was estimated as 11 months in the high-risk group, 37 months in the low-risk group, and 18 months in the intermediate-risk group. The risk classification system suggested by the joint results of the Mayo Clinic and French researchers is similar to the Spanish system, although the high-risk group is limited to complex and monosomal karyotypes and a sole der(3q) alteration is added to the low-risk group [25]. The differences in survival times based on risk groups were much larger in this report, with a median survival time of 4 months in the high-risk group, 21 months in the medium-risk group, and 41 months in the low-risk group.
Genetic characterization
An analysis of the mutational landscape provides clues to distinguishing CMML from many similar diseases, and predictions of treatment response and prognosis provided by mutations in each gene and specific molecular signatures are useful to plan treatment strategies for patients. Most CMML patients have mutations in ≥1 genes, with an average of 10–15 somatic mutations in the coding regions [27-29]. When categorized according to function, the subgroups of mutant genes that are closely related to CMML encode epigenetic regulators including those involved in DNA methylation (TET2, DNMT3A), RNA splicing (SRSF2, SF3B1, U2AF1, ZRSR2), and histone modification (ASXL1, EZH2); cell signaling pathway-related genes (NRAS, FLT3, CBL, JAK2); and a transcriptional regulator (RUNX1). Some of these gene mutations are also commonly observed in age-related clonal hematopoiesis, suggesting that the aging process of the hematopoietic system partially contributes to the pathophysiology of CMML [27].
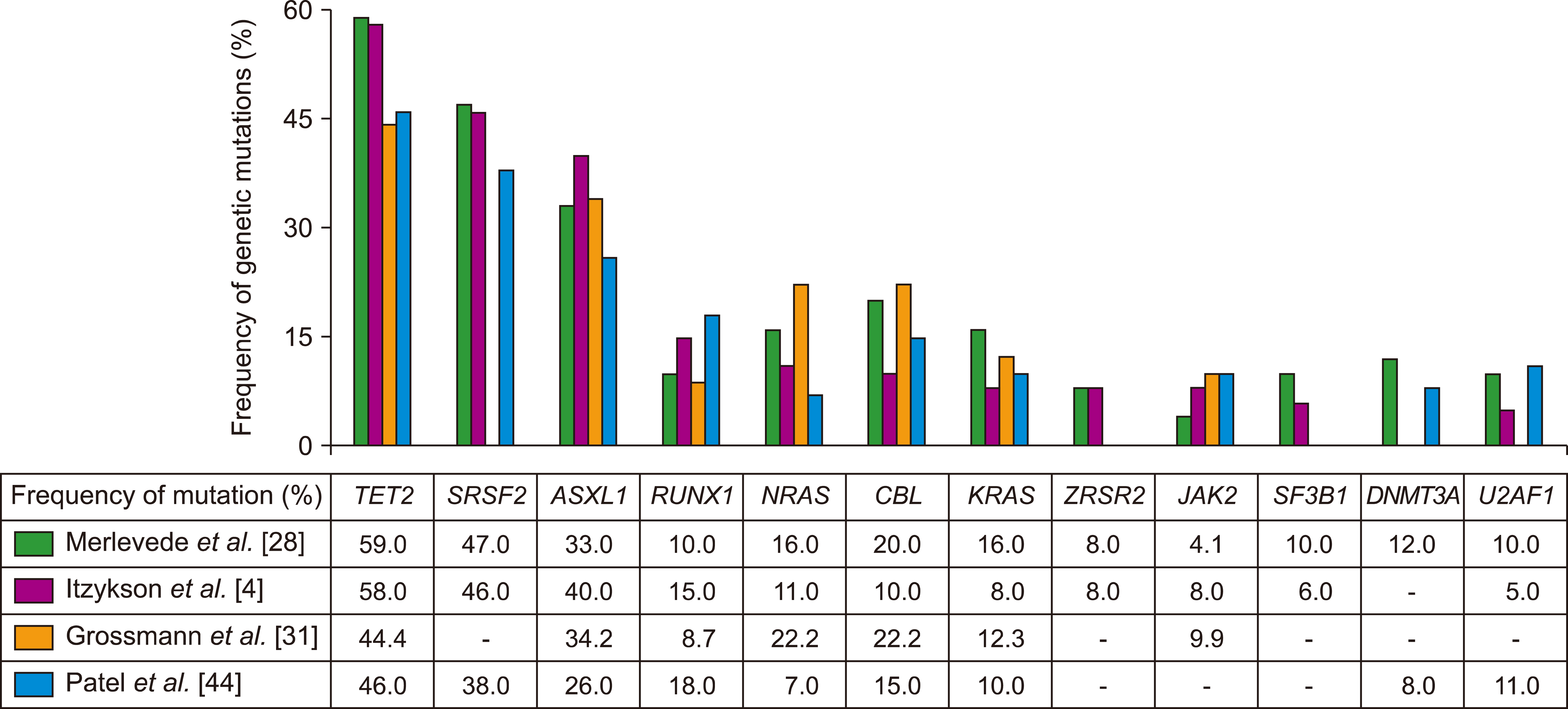
At the individual gene level, several studies have reported that SRSF2, TET2, and ASXL1 are most commonly found in CMML, each with a frequency of ≥40% (Fig. 1) [4, 27, 28, 30-32]. The co-occurrence of monocytosis and these molecular features are important in distinguishing reactive monocytosis from clonal diseases such as CMML. When the target sequencing results of 283 patients with monocytosis were previously analyzed, ≥1 gene mutations were found in 57% of 65 patients with monocytosis but without myeloid disease in the bone marrow diagnosed according to the WHO classification [33]. The most common gene mutations found in these patients were in TET2, SRSF2, and ASXL1, as seen in CMML patients. In addition, among patients not diagnosed with myeloid disease, those with mutations had an inferior prognosis relative to those without mutations, and their survival was similar to that of patients with CMML. Although the bone marrow findings are not typical, it can be assumed that the appearance of a particular genetic feature with a pattern similar to that in CMML is a way to predict the occurrence of a clonal disorder and suggests that it is a precursor to CMML.
In addition, molecular feature identification is valuable for discriminating each disease within the MDS/MPN category. In a recent study of the molecular landscape of 367 MDS/MPN patients including 119 CMML patients, CMML was more frequently accompanied by TET2, SRSF2, and KRAS genetic variants than the other subtypes. Moreover, an ancestral TET2 mutation was particularly more frequently found in the CMML subgroup than in other types of MDS/MPN [34]. Mutations in the RUNX1 gene and in signaling genes were often found as secondary findings. Through an analysis based on variant allele frequency, the authors suggested that TET2 mutation occurs in the early stages of CMML development and SRSF2 and ASXL1 mutations occur with or after TET2 alteration.
An ASXL1 variant has been identified as a gene mutation associated with the prognosis of CMML. This gene encodes a chromatin-binding protein and regulates the function of the polycomb complex PRC2 involved in histone modification, and mutated ASXL1 inhibits this regulatory function [35]. ASXL1 mutations are commonly recognized as a poor prognostic factor in myeloid neoplasms, and the recent European LeukemiaNet guidelines classify patients with AML with an ASXL1 mutation as a poor-risk group [36]. In a study by Itzykson et al. [4], CMML patients with such mutations were more anemic and often showed high WBC counts. Moreover, the ASXL1 mutation itself was found to be an independent poor prognostic factor along with old age and subpar hematologic parameters. Patnaik et al. [5, 37] reported that nonsense or frameshift mutations in ASXL1 are a prognostic factor suggestive of a low survival rate. In addition, an ASXL1 mutation leads to a high risk of transformation to AML and is associated with a low survival rate [30]. Mutations in EZH2, another factor involved in ASXL1-related leukemogenesis, are often found concomitant with mutations in ASXL1. Co-mutation of ASXL1 and EZH2 is common in the CMML-2 type and is associated with a low survival rate; however, such mutations have no prognostic relevance by themselves [37]. On the basis of these findings, many researchers have recently added ASXL1 mutations as a variable to the previously proposed cytogenetic-based risk prediction model [5, 6].
With respect to genetic mutations other than ASXL1, their prognostic relevance has not yet been clearly identified. TET2 mutations are known to be associated with better survival if they occur together with wild-type ASXL1 [38]. SFTBP1 mutations are associated with a low survival rate, but are rarely observed in CMML [39]. DNMT3A mutations are also not common in CMML but are believed to be associated with low survival rates and rapid evolution to AML [40]. The RUNX1 mutation is not associated with survival rate but is assumed to contribute to leukemic transformation [41]. As studies on these genes have heterogeneous characteristics and varying numbers of the patient population, in addition to reporting inconsistent results, these genes have not yet been incorporated into diagnostic criteria or risk prediction systems. To clarify the exact function of each gene in CMML, additional evidence needs to be accumulated.
Mutations in genes belonging to the RAS signaling pathway, including NRAS, KRAS, and CBL, are also relatively common in CMML. RAS-related gene mutations are more closely related to the proliferative phenotype of CMML and have been reported to have a negative impact on prognosis [12, 30, 42-45]. However, this poor prognostic impact of RAS pathway alterations does not rule out the possibility that the proliferative phenotype of the mutant group and concomitant mutation in high-risk genes such as ASXL1 are affected. In a large-scale retrospective study, it was found that biallelic mutations in TET2 and mutations in genes belonging to the RAS family are associated with progression in CMML [44]. Mutations in genes of the RAS pathway are often found in the nodules of plasmacytoid dendritic cells observed in the bone marrow of some CMML patients, and it has also been reported that these mutations are associated with a high frequency of transformation to AML [46]. Recently, experimental evidence has indicated that mutations in NRAS that occur with Tet2 loss cause transformation to AML [47]. In summary, although the prognostic effect of mutations in RAS-associated genes themselves on CMML is not yet clear, molecular alterations to RAS pathway genes in CMML are believed to contribute to the development and evolution of this disease through certain mechanisms, such as the induction of uncontrolled hematopoietic cell proliferation.
RISK STRATIFICATION
Risk stratification is important to predict disease evolution and mortality in CMML patients; however, it is sometimes difficult owing to the heterogeneous nature of the patient population. Several research groups have published criteria for classifying CMML risk groups, although most of the recommendations were based on the results of retrospective analyses. Further, the disease-related or patient-related factors used in risk estimation slightly differ across the classification systems.
The approximate contents of the various classification systems proposed to date are summarized in Table 2. They commonly adopt low hemoglobin level as a risk factor, and the CMML-specific prognostic scoring system (CPSS) only includes red blood cell (RBC) transfusion dependence instead of hemoglobin level [24]. Increased WBC counts, especially monocytes and lymphocytes, are associated with a poor prognosis, and thrombocytopenia is also included in many classification systems. Increased blasts in the bone marrow and immature myeloid cells appearing in peripheral blood are also factors that add to the risk stratification. Among the patient-related factors, age and performance status have been included in the global MD Anderson prognostic score (MDAPS) classification [48].
Cytogenetic abnormality is an important consideration in risk estimation. Kantarjian et al. [48] determined that patients with chromosome 7 abnormalities and a complex karyotype are a high risk group. In the CPSS, patients with trisomy 8, a complex karyotype (≥3 abnormalities), and chromosome 7 abnormalities are classified as a high-risk group; those with normal and isolated –Y are classified as a low-risk group; and those with other abnormalities are classified as an intermediate-risk group. These groups are assigned 2 points, 0 point, and 1 point, respectively [24].
Recently, as it is known that mutations in specific genes affect prognosis, new classification criteria that added genetic factors to the existing prognostic scoring system were announced. At the Mayo Clinic, Patnaik et al. [49] analyzed the prognostic significance of the ASXL1 mutation and found that the significance disappeared in multivariate analysis, leading to the exclusion of this mutation from the scoring system. However, of the many types of ASXL1 mutations, only nonsense and frameshift mutations were found to be associated with prognosis. Accordingly, a Mayo molecular model using 5 variables (anemia, thrombocytopenia, increased absolute monocytic count, circulating immature myeloid cells, and nonsense/frameshift mutations of ASXL1) was proposed [5]. In the Groupe Francophone des Myélodysplasies scoring system published by Itzykson et al. [4], nonsense/frameshift mutations of ASXL1 were added as risk factors along with old age, anemia, thrombocytopenia, and leukocytosis. In the latest updated CPSS published in 2016, scores according to the genetic scoring system using the karyotype and mutations in ASXL1, NRAS, RUNX1, and SETBP1 were incorporated into the existing CPSS [6].
Taken together, to assess the overall risk of CMML patients, peripheral blood test, bone marrow evaluation, and chromosomal analysis are essential, and mutation tests of major genes are also required. Therefore, these facts should be considered in the initial workup of CMML patients. However, despite numerous studies, there is still no risk scoring system that can be commonly applied. This is probably because of the relatively small number and heterogeneity of CMML patients. Moreover, variables related to comorbid conditions other than CMML may also influence the risk assessment of CMML patients, who are mostly elderly. The existing CMML risk scoring systems can be reorganized and individually applied in various ways according to the circumstances of each institution. In addition, further studies and data accumulation are needed.
TREATMENT
Determining which patients need treatment
CMML is a group of diseases with heterogeneous characteristics, and the clinical course and prognosis differ among patients. After a CMML diagnosis, the primary consideration is determining whether the patient needs treatment followed by deciding the appropriate treatment. The establishment of a plan for long-term treatment must be preceded by an appropriate risk classification.
Patients with no symptoms and who have a low risk, as well as those with disease classified as CMML-0 according to the WHO classification, have a relatively stable and indolent disease course and a generally low probability of progression to AML. These patients may be followed up until evidence of disease progression is found or until clinically significant symptoms develop [7, 8]. Through this approach, asymptomatic CMML patients with a low risk can avoid treatment complications and the concomitant deterioration in their quality of life. However, for high-risk patients, the disease course needs to be modified to prevent disease evolution or death. A definite treatment aimed at cure, including allogeneic HSCT, is recommended for young and healthy patients with a high risk or severe clinical symptoms [7-9]. Regardless of whether a transplant is scheduled, HMAs are usually the preferred treatment to reduce the leukemic burden and overcome transfusion dependence in patients who need treatment. In addition, hydroxyurea for cytoreduction is still used.
HMAs
HMAs, including azacitidine and decitabine, induce hypomethylation by inhibiting DNA methyltransferase (as a representative mechanism of action) and are widely used for myeloid neoplasms. In the Republic of Korea and the United States, both azacitidine and decitabine are approved for use for MDS including CMML, whereas only azacitidine is licensed in Europe.
The clinical trials that led to the approval of these drugs were conducted on a large number of MDS patients, and the overall response rate (ORR) reached 60% [50, 51]. As the number of CMML patients enrolled in these studies was small (<20), the data are insufficient to accurately evaluate the effect of HMAs on CMML. The results of a phase III clinical trial limited to CMML patients have not yet been reported. In phase II clinical trials in which only CMML patients were enrolled, an ORR of approximately 30–50% and a complete response rate of 10–20% were reported, although the number of patients was small [52-55]. In addition, many retrospective studies have analyzed various numbers of CMML patients treated with HMAs, reporting widely ranging ORRs of 25–75% [56-66]. In addition, Subari et al. [67] reported that HMA induces spleen size reduction at a rate of 45% in cases of CMML with splenomegaly. On the basis of the aforementioned results, despite the limited available data, HMA treatment for CMML can be expected to result in more than a moderate bone marrow (hematologic) response and an improvement in the clinical symptoms of patients.
However, it is difficult to select candidates for HMA treatment among CMML patients because it is not yet clear which factors predict the response to such therapies. Some of the several retrospective studies have identified clinical factors associated with response rates; however, the results of those studies were inconsistent. In a retrospective analysis of 174 patients, Duchmann et al. [65] showed that ASXL1 mutations were associated with a low HMA treatment response rate and that a TET2 mutation without an ASXL1 mutation was a predictor of high rates of complete remission and low rates of mortality. In contrast, in a retrospective analysis of HMA treatment results in 121 CMML patients conducted by Costa et al. [58], only serum lactate dehydrogenase level was a predictor of response, whereas genetic factors including ASXL1 and TET2 mutations had no effects. In another study, the CMML subtype classified according to the WHO classification and the response rate were correlated [64]. However, no predictors of response proved to be significant in phase II clinical trials [52, 53, 55]. Therefore, at present, the application of HMA treatment to CMML patients should be individualized considering various factors, including the severity of clinical symptoms, the risk according to the scoring system, and the patient’s medical fitness.
Drugs that can replace conventional HMAs, including subcutaneous azacitidine and intravenous decitabine, have recently been developed. A phase I clinical trial of oral azacitidine published in 2011 included 45 patients with myeloid neoplasms, of whom 4 had CMML [68]. In this study, oral azacitidine as a frontline therapy showed a high ORR of 73%; however, owing to the small number of patients, the therapeutic effect against CMML alone could not be judged. Recently, an oral combination of cedazuridine and decitabine was introduced. Cedazuridine is an inhibitor of cytidine deaminase that, when administered with the HMA decitabine, prevents the destruction of decitabine and increases its bioavailability [69]. Oral cedazuridine/decitabine achieved an ORR of 60% in a phase II clinical trial conducted in patients with MDS and CMML, as well as a safety profile and clinical activity similar to those of intravenous decitabine in a phase III clinical trial [70, 71]. On the basis of these results, oral cedazuridine/decitabine was approved as a therapeutic agent for untreated, intermediate 1/2, or high-risk MDS and CMML in the United States and Canada [72].
Allogeneic HSCT
Allogeneic HSCT remains the only curative option for CMML patients, although there are still problems such as the high treatment-related mortality and difficulty in the identification of suitable donors. Moreover, there is little information on the selection criteria for patients requiring transplantation, standard therapy for pre-transplant treatment, intensity of conditioning chemotherapy for transplantation, frequency of toxicity, and final outcomes. Several researchers have retrospectively analyzed and reported the results of allogeneic HSCT for CMML patients [73-80]. In their studies, allogeneic HSCT had a response rate of 20–40%, a recurrence rate of 20–40%, and an overall 5-year survival rate of approximately 20%. These studies included a small number of patients (range, 12–85 patients) and varied in the clinical characteristics of the patients and the types of treatments received before HSCT. Nevertheless, it has been found that the prognostic factors associated with the prognosis of transplantation include cytogenetic characteristics [78, 79], percentage of blasts in the bone marrow [78], age [79], and comorbidities [74, 79]. In particular, patients who were classified into the high-risk group according to the prognostic scoring system based on karyotype were reported to have inferior prognosis after transplantation [79]. This result suggests that risk stratification and grouping using an appropriate prognostic model at the first diagnosis of CMML is important for the overall clinical course, including the treatment decision and disease outcome. As mentioned in the previous section, in addition to the karyotype, recent prognostic scoring systems include genetic factors. As there is still no information about the effects of mutations in specific genes on transplant outcomes, continuous research is needed.
The first treatment considered for CMML, especially for high-risk groups, is HMAs. However, intensive chemotherapy to clear malignant cells may be another option when a transplant is planned. A retrospective study that analyzed a large group of 513 patients showed that achieving complete remission at the time of HSCT was the only significant predictive factor of relapse-free and overall survival [81]. However, as CMML patients are often elderly and have many comorbid diseases, intensive chemotherapy is not expected to yield good results owing to safety issues. Kongtim et al. [82] reported that patients treated with an HMA before transplantation had superior response, treatment-related mortality, and progression-free survival rates relative to those in the intensive chemotherapy group, which is probably attributable to the less concern for toxicity with cytotoxic chemotherapy before transplantation. In addition, Sekeres et al. [83] reported the outcome of allogeneic HSCT after azacitidine-based low-intensity treatment in 277 high-risk MDS and CMML patients, who showed similar outcome to that of historical controls who received transplantation after intensive chemotherapy. Although no standard pre-transplantation treatment has been established, a treatment designed to maximize the pre-transplantation marrow responses while minimizing toxicity should be selected. In this process, it is necessary to consider the characteristics of the disease itself (e.g., karyotype), as well as factors related to the general health status and comorbidity of the patients (e.g., age and performance status). These considerations contribute to the individualization of the patients’ treatment.
Cytoreduction and supportive care
Cytoreductive therapy for the treatment of leukocytosis and organomegaly associated with CMML, as well as various constitutional symptoms, has been considered an important component of CMML treatment, despite the lack of a cure for the disease. For cytoreductive therapy, low-dose cytotoxic agents, including cytarabine, etoposide, and hydroxyurea, are mainly selected. Among them, hydroxyurea is the most widely used drug to date. Hydroxyurea was demonstrated to be superior in terms of both response rate and survival relative to etoposide in a randomized trial in CMML patients [84]. In addition to the important clinical benefit, hydroxyurea is an oral drug, thus guaranteeing ease of use. Therefore, it is preferentially recommended as a symptomatic treatment for patients who are not receiving intensive treatment and as a bridge therapy before HMAs [7, 8].
To improve anemia in MDS and overcome transfusion dependence, an erythropoietin-stimulating agent (ESA) is primarily considered. The effect of ESA in patients with low-risk MDS with a low erythropoietin level is well known [85-87]. In a study in 94 CMML patients, ESA treatment resulted in an erythroid response rate of 64% and a transfusion independence rate of 31% [88]. Therefore, as for other MDS, the use of ESA is recommended for symptomatic anemia among patients who are not receiving definite treatment such as HMAs or allogeneic HSCT. RBC transfusion is the only option for anemia that is refractory to ESA or other treatments. However, repetitive RBC transfusion carries the risk of iron overload. Similar to MDS patients, CMML patients with transfusion dependence needs iron chelation therapy [7, 8].
Novel therapeutic agents
In addition to the previously used cytotoxic agents and transplantation methods, new therapeutic drugs targeting the unique characteristics of hematopoietic cells, various signals contributing to malignant transformation and proliferation, and specific mutations and surface markers are continuously being developed. Most of the new drugs under development aim to target several myeloid neoplasms, including MDS, rather than CMML alone (Table 3). Currently, the new drugs with therapeutic potential for myeloid neoplasms including CMML are Janus kinase 2 inhibitors (ruxolitinib and pacritinib) [89, 90], RAS/mitogen-activated protein kinase signaling pathway inhibitors (tipifarnib and trametinib) [91-93], recombinant fusion protein with interleukin-3 and toxin [tagraxofusp (formerly SL-401)] [94], anti-granulocyte–macrophage colony-stimulating factor monoclonal antibody (lenzilumab) [95, 96], and a spliceosome inhibitor (H3B-8800) [97, 98].
As these novel agents are still limited by their low effectiveness and toxicity, their usefulness must be proven through numerous studies before they can be introduced into clinical practice. Nevertheless, it is clear that the basis of treatment for CMML is gradually widening.
CONCLUSION
Despite much research and the continuous accumulation of knowledge, many aspects of CMML remain unknown. In addition to the existing diagnostic criteria that depend on morphologic findings, novel data obtained by recent techniques, including MPFC and next-generation sequencing, facilitate the differential diagnoses of diseases accompanying monocytosis and are also used to understand and predict the natural course of CMML. Despite the availability of several therapeutic agents, the treatment of CMML is still not satisfactory. To properly apply various treatments, patients should be approached individually based on an accurate understanding of clinical and disease factors. Moreover, owing to the lack of standard treatment strategies, it is reasonable to actively consider possible clinical trial participation when treating patients with CMML. The current knowledge about CMML had been extracted from studies conducted with a small number of CMML patients, most of them included as part of MDS cohorts. In the future, research and clinical trials reflecting the characteristics of CMML patients should be continuously conducted in both the diagnosis and treatment aspects.
 XML Download
XML Download