

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Myeloproliferative neoplasms (MPNs) are clonal disorders of hematopoietic stem cells (HSCs), in which one or more myeloid lineages are involved [1]. In typical Philadelphia chromosome (Ph)-negative MPNs, which include polycythemia vera (PV), primary myelofibrosis (PMF), and essential thrombocythemia (ET), the majority of patients harbor mutations in the Janus kinase 2 (JAK2), calreticulin (CALR), or thrombopoietin receptor (MPL), which drive clonal HSC expansion [2-4]. Typical Ph-negative MPNs manifest a broad spectrum of clinical presentations ranging from asymptomatic to limited constitutional symptoms, such as fatigue, pruritus, and general weakness, as well as an increased risk of thrombosis and progression to leukemia.
The goals of ET and PV treatment are thrombosis prevention, control of general symptoms, and monitoring of the progression to leukemia. Antiplatelet agents, including low-dose acetylsalicylic acid and cytoreductive agents, such as hydroxyurea and anagrelide, are mainly used [5]. In PMF, ruxolitinib has shown improvement in symptoms and a decrease in splenomegaly and is associated with a gain in survival compared to conventional therapy [6, 7]. However, allogeneic hematopoietic stem-cell transplantation is currently the only potentially curative therapy [8, 9], and limited treatment options are available.
Interferon alpha (IFNα) was the first immunotherapeutic agent approved by the Food and Drug Administration (FDA) in 1986 for clinical use in cancer [10]. It has been used in the treatment of MPNs for approximately 40 years. IFNα was first identified almost 60 years ago for its antiviral activity [11]. In addition to antiviral effects, it also plays a role in anti-proliferative, pro-apoptotic, and immunomodulatory responses. IFNα is an attractive option for the treatment of MPN and induces symptom control, a hematologic response, and disease-modifying activity. However, its frequent dosage and toxicity profile are major barriers to its widespread use. The development of more tolerable forms of IFNα, including pegylated IFNα (peg-IFNα), and more recently, ropeginterferon alpha-2b (ropeg-IFNα-2b), has further increased interest in this therapy [12-14]. The purpose of this article is to discuss the current and future role of IFNα in the treatment of Ph-negative MPNs through a review of the latest papers.
Background for the treatment of MPN with IFNα
MPNs are inflammatory cancers, wherein the malignant clone triggers inflammatory cytokines, which sustain the inflammatory drive in a self-perpetuating vicious cycle. Disease progression is in the biological continuum from the early stages of cancer, such as ET and PV, to the advanced myelofibrosis stage and impending leukemic transformation [12]. Additional mutations, aside from the drive mutations (JAK2, CALR, and MPL), emerge during this evolution [15].
Hydroxyurea is generally accepted as the first-line therapy for high-risk patients with PV and ET because of its ease of administration and low cost [16]. Since a significant number of patients are either intolerant of hydroxyurea because of hematologic or non-hematologic toxicity or resistance due to a lack of effective cytoreduction, there is a need for alternative therapeutic agents. In addition, there are concerns about secondary malignancies or leukemic transformation when using hydroxyurea for a prolonged duration [17, 18]. Although IFNα is currently not approved by the FDA for the treatment of ET and PV, consensus guidelines recommend IFNα as an option for first-line cytoreductive therapy, particularly in younger or pregnant patients [19]. IFNα controls myeloid cell increase, reduces spleen size, and provides relief from related symptoms.
JAK inhibitors are primarily used in the treatment of symptomatic patients with high-risk MF [6, 20]. Although the recent approval of the JAK inhibitors ruxolitinib and fedratinib has significantly improved the management of symptoms in patients with MF, a considerable proportion of patients are either refractory to ruxolitinib therapy or experience dose- or treatment-limiting adverse effects. The optimal treatment of patients with low-risk MF and those who are intolerant or refractory to JAK inhibitors continues to evolve. Several studies have demonstrated that IFNα results in significantly decreased bone marrow fibrosis in patients with MF [21, 22].
IFNα belongs to a large class of proteins known as cytokines and is among molecules used for communication between cells to trigger the protective defenses of the immune system that help to eradicate viruses [23]. It exhibits significant antiviral effects, influences the quality of the cellular immune responses, and amplifies antigen presentation to specific T cells by increasing the expression of major histocompatibility complex antigens [24]. IFNα can also suppress angiogenesis by downregulating angiogenic stimuli derived from tumor cells and control the proliferation of endothelial cells. Such suppression decreases tumor angiogenesis and vascularization and subsequently inhibits tumor growth [25, 26].
An initial study described IFNα as an effective treatment for controlling thrombocytosis in MPNs [27, 28]. Since then, several studies have confirmed that IFNα can also inhibit myeloproliferation in Ph-negative MPNs, reduce the need for phlebotomies in PV, provide relief from pruritus, normalize elevated leukocyte and platelet counts, and reduce spleen size [12-14]. Despite these advantages, IFNα is not the first drug of choice for the treatment of MPNs because of its relatively high discontinuation rate due to adverse effects. Only recently has interest in IFNα re-emerged. With the identification of the JAK2V617F mutation in 2005 [29], there have been reports about the potential of IFNα to induce molecular remission in JAK2V617F-positive patients [30, 31]. Following the discovery of CALR mutations in 2013 [3], a reduction in the load of these mutations and the treatment of CALR-positive MPN patients with IFNα has been reviewed in recent years [32, 33]. In some patients, the molecular response was maintained by long-term treatment with IFNα [34]. These findings show that treatment with IFNα has the potential for disease modification in some patients with MPNs.
Review of recent IFNα data in PV and ET
Bewersdorf et al. [35] conducted a systematic review and meta-analysis screening of all studies on effect of IFNα, in PV and ET, conducted until March 2019. In total, 44 studies with 1,359 patients (730 ET and 629 PV) were included. The overall response rate (ORR) was defined as a composite of the complete hematologic response (CHR) and partial hematologic response (PHR). The ORR was 80.6% (CHR 59.0%) and 76.7% (CHR 48.5%) for ET and PV patients, respectively. However, despite its promising therapeutic potential, the adverse effect profile of IFNα and frequent subcutaneous dosage have been major reasons for its disuse in recent decades. Given these limitations, pegylated forms of IFN have been developed, which have better tolerability and can be administered once a week. Clinical studies have demonstrated the effectiveness of peg-IFNα in a large number of patients with Ph-negative MPNs. Several studies have shown that peg-IFNα can reduce the burden of the JAK2V617F mutation, which suggests a disease-modifying effect that is uncommon with hydroxyurea [31, 36]. Notably, the presence of concomitant non-driver mutations was associated with a lower mean decrease in the burden of the JAK2V617F mutation [37].
The MPD-RC 111 trial evaluated the response to peg-IFNα therapy in patients with ET and PV who had previously been treated with hydroxyurea (Table 1) [38]. The ORR [complete response (CR)+partial response (PR)] at 12 months was 69.2% (CR 43.1%) and 60% (CR 22%) in patients with ET and PV, respectively. The CR rates were significantly higher in ET patients with CALR mutations (56.5% vs. 28.0%; P=0.01). The MPD-RC 112 trial compared peg-IFNα to hydroxyurea in treatment-naïve patients with high-risk PV and ET [39]. At 12 months, the ORR was 69.8% and 78% for hydroxyurea and peg-IFNα treatment, respectively (P=0.22). At 24 months, the ORR was 40.7% and 59.6% for hydroxyurea and peg-IFNα treatment, respectively (P=0.04). However, peg-IFNα treatment was associated with a higher rate of grade 3/4 toxicity. Although peg-IFNα has improved tolerability compared to standard IFNα, many patients still experience adverse effects.
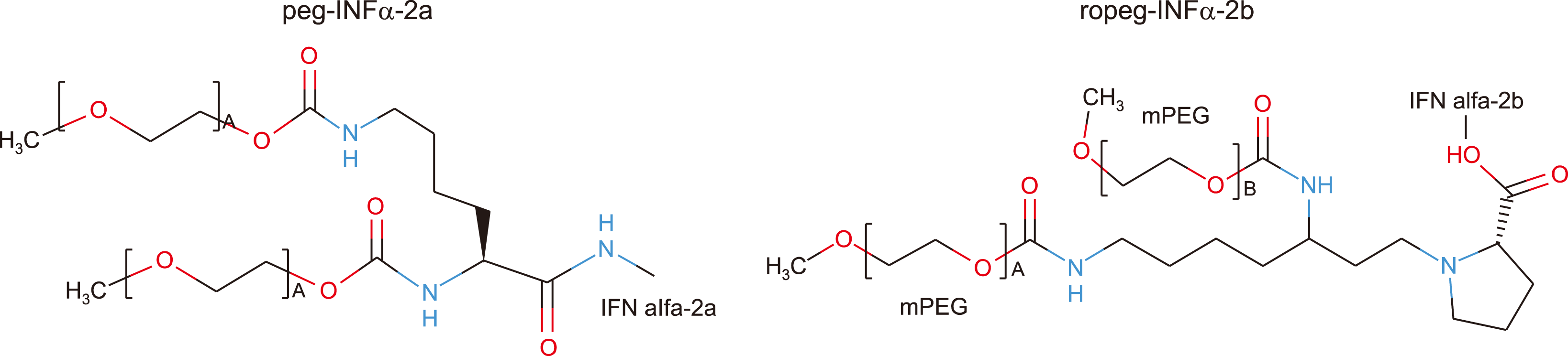
Ropeg-IFNα-2b is a structurally novel and monopegylated IFNα that is administered subcutaneously. Monopegylation allows for an extended dosing interval to every other week, improving patient compliance and tolerability, as well as decreasing fluctuations in uptake and elimination (Fig. 1). The studies PROUD-PV and its continuation CONTINUATION-PV, which enrolled 257 patients with early stage PV, have recently been published [40]. The CHR in the ropeg-IFNα-2b and standard therapy groups was 43% and 46% (P =0.63) at 12 months, respectively, in the PROUD-PV study, and it was 71% and 51% (P =0.012) at 36 months, respectively, in the CONTINUATION-PV study. In addition, the mutant JAK2 allele burden showed a rebound in the second year of hydroxyurea treatment and returned to baseline levels in the third year. In contrast, the ropeg-IFNα-2b molecular responses increased further, and the allele burden decreased to half of the baseline levels in the second and third years of treatment. The proportion of grade 3/4 toxicity was similar in both treatment groups. Based on these studies, ropeg-IFNα-2b was approved by the European Medicines Agency as a monotherapy in adults for the treatment of PV without symptomatic splenomegaly in February 2019. FDA approval remains in progress. In the MPD-RC 112, PROUD-PV, and CONTINUATION PV studies, it can be seen that when IFNα was used for >1 year, the effectiveness of treatment was better than that of hydroxyurea. Thus, it can be assumed that a considerable duration of treatment is necessary to achieve sufficient IFNα effects.
In low-risk PV patients, defined as those aged ≤60 years with no history of thrombosis, phlebotomy is currently the standard treatment to maintain the hematocrit at normal levels [16, 19]. Treatment with ropeg-IFNα-2b showed a larger proportion of patients with low-risk PV maintaining their hematocrit levels at ≤45% for a year compared to phlebotomy alone, according to the results of an interim analysis from the Low-PV trial [41]. Eighty-four percent of patients in the ropeg-IFNα-2b arm achieved the primary composite endpoint of maintaining hematocrit levels at ≤45% for 12 months in the absence of progressive disease compared to 60% in the phlebotomy arm (odds ratio, 3.5; P=0.008). No significant difference was noted in severe adverse events between the two arms. In the future, IFNα therapy should be considered for patients with early stage MPNs.
Review of recent IFNα data in MF
Bewersdorf et al. [42] conducted a systematic review and meta-analysis screening of all IFNα studies in patients with MF until March 2019. A total of 10 studies with 141 patients with MF were included. The ORR was 49.9%. Disease-modifying effects have also been reported in studies on MF, although complete molecular responses are rare. The ORR was lower in MF patients than in PV and ET patients [35, 42]. In contrast to PV and ET, MF is a far more advanced disease with additional non-driver mutations [15]. Some studies have reported that the presence of additional non-driver mutations affects the response to IFNα treatment [37]. In addition, toxicity such as cytopenia is more common in advanced MF than in PV and ET, leading to more frequent discontinuation. IFNα is recommended as an option in symptomatic low- or intermediate-risk MF patients by the National Comprehensive Cancer Network and the Nordic MPN study group [43, 44]. The successful treatment of 30 patients with early MF with IFNα was reported by Silver et al. [45]. Seventy-three percent of patients improved or remained stable with acceptable toxicity, including 37% who achieved CR or PR.
Combinations with other medications are being developed to enhance the inadequate effect of IFNα in patients with MF. Combination therapies with potent anti-inflammatory agents, such as the JAK inhibitor ruxolitinib, can potentially enhance IFNα signaling, given that high levels of inflammation may be involved in IFNα resistance. Combination therapy may also permit lower doses, thus improving tolerability. The COMBI trial evaluated ruxolitinib and peg-IFNα in 32 PV and 18 MF patients [46]. There were no high-risk MF patients included in this trial, with the majority being low-risk (N=6) or intermediate-1 risk (N=9) according to the Dynamic International Prognosis Scoring System-Plus score. Forty-four percent of patients with MF achieved remission. The median JAK2V617F allele burden decreased from 47% to 12%, and 41% of the patients achieved a molecular response. Discontinuation rates were observed in 32% of MF patients.
Administration of IFNα
IFNα treatment is associated with adverse events that account for discontinuation rates of approximately 20–30% in most studies [12, 31, 39]. Adverse effects of IFNα have been described in almost every organ system, and many of them are dose-dependent [47, 48]. Flu-like symptoms (40–60%), hematological toxicity (20–30%), elevated transaminase levels (20–30%), nausea, fatigue, and psychiatric sequelae are the most frequently encountered adverse effects. Many patients experience initial flu-like symptoms, such as fever, myalgia, and chills. This can be reduced by pre-medication with corticosteroids, ibuprofen, and acetaminophen and by administering the IFNα dose at night [49]. Flu-like symptoms usually subside with repeated dosing but often recur with each dose increase; hence, the dose should only be increased once tolerability at each dosage has been confirmed. In some patients, chronic fatigue and/or musculoskeletal pain may persist, ultimately necessitating treatment withdrawal. Some patients may develop symptoms and signs of autoimmune diseases. Thyroid dysfunction, or thyroiditis with ensuing hypothyroidism, may develop in a subset of patients. Thyroid function tests should be performed at baseline and at least annually during the treatment, although more frequent monitoring may be needed for female patients during the first year of therapy [50]. Other rare autoimmune diseases that may be exacerbated by IFNα include polyarthritis, dermatomyositis, immune hemolytic anemia, immune thrombocytopenia, and glomerulonephritis [51]. A history of depression is a relative contraindication for IFNα therapy. Depression, suicidal ideation, and attempted suicide have been reported during treatment and within 6 months following discontinuation. Patients should be evaluated for signs or symptoms of mood disorders, and if depression develops or worsens, IFNα should be discontinued, and psychiatric intervention should be provided as appropriate [52, 53]. Appropriate patient selection, education, and the proper support of an experienced hematology clinical team can assist in reducing the rate of discontinuation due to side effects.
Although patients with MPNs may achieve CHR or molecular response with IFNα, lifelong treatment constitutes a major burden for these patients. There are no studies on the discontinuation of IFNα when the treatment response is good. An abstract of a study was presented at the American Society of Hematology in 2020 [54]. A total of 381 MPN patients treated with IFNα were included in the study. After a median follow-up of 72.4 months from IFNα initiation, 131 patients were still on IFNα treatment, while 250 patients discontinued therapy. The reasons for discontinuation were toxicity in 128 patients (50.4%), prolonged hematological CHR in 76 patients (29.9%), response failure in 16 patients (6.3%), and other reasons in 30 patients (11.8%). At the time of IFNα discontinuation, 170 patients (66.9%) displayed CHR, and the variant allele frequency of the median driver mutation was 12%. Of note, IFNα was re-introduced in 61 patients who did not maintain CHR. The rate of CHR during the second treatment was 83.6%, suggesting no resistance to IFNα in post-discontinuation relapses. Although follow-up or additional studies are necessary, it is thought that IFNα discontinuation could be considered in MPN patients who have achieved CHR.
Future directions of IFNα treatment
IFNα is an effective drug that can modify the course of the disease and control the symptoms through cytoreduction. In particular, the effect of ropeg-IFNα-2b, which is known to have fewer side effects, is expected to be beneficial. If the FDA approval of ropeg-IFNα-2b for use in PV patients is granted, it is expected to be used more widely. If the adverse effects can be controlled, IFNα can be used not only to replace hydroxyurea for cytoreductive therapy in high-risk patients but also for symptomatic control in low-risk patients.
Immunotherapeutic approaches are expanding and will ideally extend the therapeutic modality in patients with MPNs. IFNα is a non-specific immunotherapy, and there are limitations to monotherapy. Further clinical studies are necessary to investigate the effect of combination therapy with IFNα and other drugs to overcome these limitations. Combination therapy with IFNα and ruxolitinib has already been studied. Beyond JAK inhibitors, multiple new agents are being investigated as potential combination drugs for patients with MF [55].
CONCLUSION
IFNα is the treatment of choice with potential for disease modification, given its impact on mutation burden and achievement of a durable response. However, its administration and toxicity profile are major barriers to its widespread use, which has led to the development of peg-IFN, and more recently, ropeg-IFNα-2b. In PV and ET, IFNα is expected to be used as the first-line therapy for cytoreductive treatment, as well as in patients with hydroxyurea intolerance and resistance. In MF, a good result from combination therapies with IFNα and other drugs, such as ruxolitinib, is expected.
 XML Download
XML Download