

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Inherited hemoglobin (Hb) disorders are classified into two disease groups: hemoglobinopathy caused by the structural defects in Hb and thalassemia caused by the impaired synthesis of globin, usually of normal structure; both are typically inherited in an autosomal recessive manner [1]. It has been estimated that 70,000 children are born with various forms of thalassemia annually [2]. The term “thalassemia” is derived from the Greek word “thalassa (sea),” as this inherited disease was first described in the Mediterranean region. Thalassemia is also highly prevalent in other regions of the world (e.g., Southeast Asia, the Indian subcontinent, and Africa), where malaria is endemic. Moreover, the prevalence of thalassemia has increased in recent years, due to an increase in migrant populations in other regions, including North America, Northern Europe, Australia, and South Korea. It is estimated that approximately 7% of the global population are thalassemia carriers [3].
Thalassemia is classified into α-, β-, δβ-, γδβ-, δ-, and γ-thalassemia, which are identified by the specific type of globin chain(s) where impaired synthesis occurs. For example, α- and β-thalassemia are deficient in the synthesis of α- and β-globin chains, respectively [4]. Among these various forms, the two most common forms are α- and β-thalassemia [5]. This review describes the genetic basis and molecular diagnosis of thalassemia, focusing on α- and β-thalassemia.
GENETIC BASIS OF HEMOGLOBIN
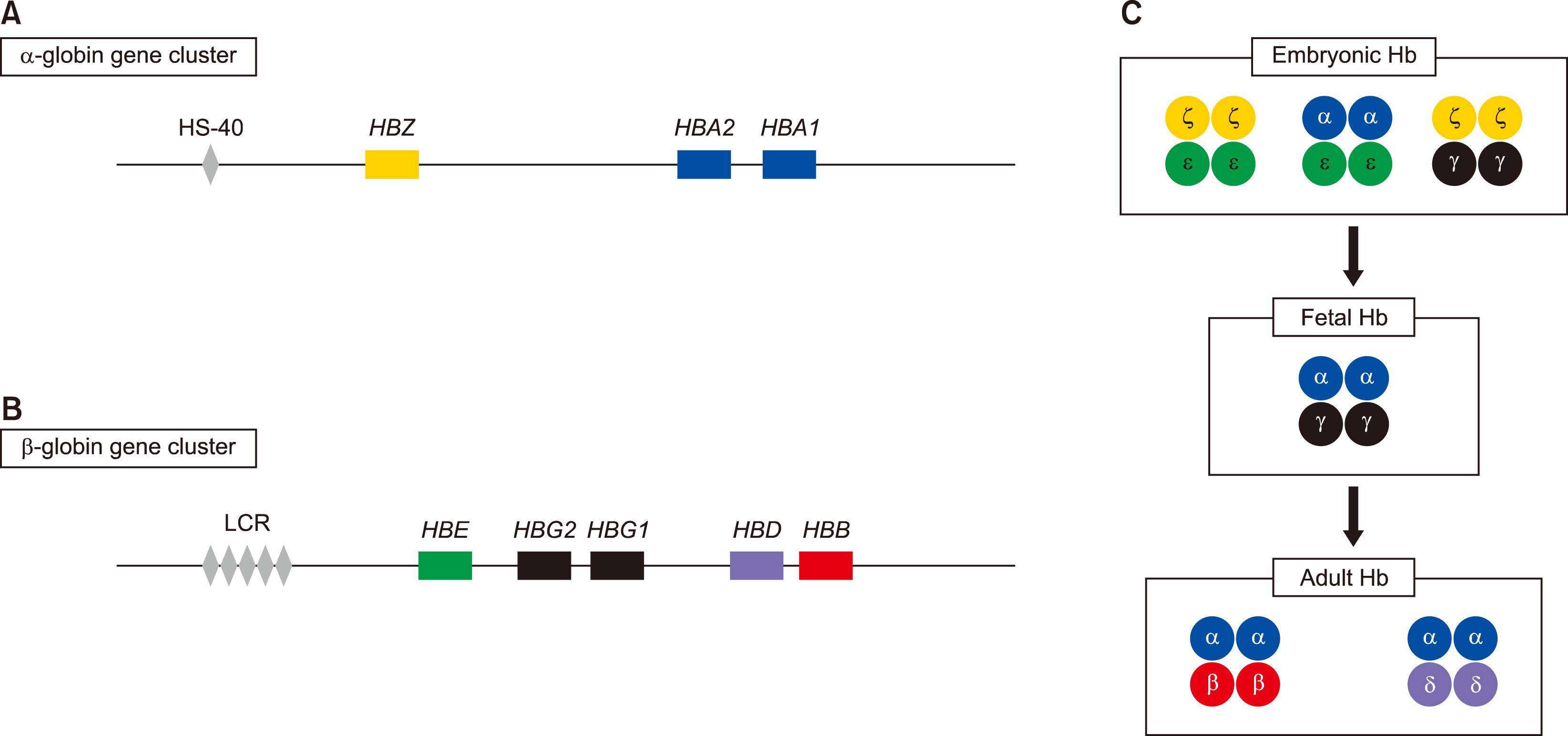
Human Hb consists of proteins with symmetric pairing of α-like and β-like globin dimers, which form a tetrameric structure, as well as functional units [6]. Individual α-like and β-like globin chains are encoded by two distinct gene clusters: the α-globin gene cluster on the short arm of chromosome 16 and the β-globin gene cluster on the short arm of chromosome 11. The α-globin gene cluster comprises three functional globin genes, the embryonic ζ gene (HBZ), and two fetal/adult α (α1 and α2) genes (HBA1 and HBA2) (Fig. 1A). The β-globin gene cluster contains five functional genes, the embryonic ε gene (HBE), two fetal Gγ and Aγ genes (HBG2 and HBG1), and adult δ and β (HBD and HBB) genes (Fig. 1B). These genes are arranged along each chromosome, and are differentially expressed at each stage of development to produce different Hb tetramers [7]. The embryonic Hb includes Hb Portland (ζ2γ2), Hb Gower-1 (ζ2ε2), and Hb Gower-2 (α2ε2), and the fetal Hb consists of α2γ2. In adults, Hb A (α2β2) accounts for 95% of the total Hb, while Hb A2 (α2δ2) constitutes the remaining 5% (Fig. 1C) [5]. The upstream region of each α-globin and β-globin gene cluster contains cis-acting regulatory elements that play a role in the regulation of globin gene expression. Within 30–70 kb upstream of the α-globin gene cluster, multispecies conserved sequence (MCS) regions (MCS-R1, 2, 3, and 4) were found. MCR-R2, also known as HS-40, is a single DNase hypersensitive site that is crucial for α-globin gene expression [5]. β-globin gene expression is regulated by the locus control region (LCR), which consists of five DNase I hypersensitive sites (HS-1, 2, 3, 4, and 5). β-globin LCR (β-LCR) spans 34 kb upstream of the ε-globin gene.
MOLECULAR BASIS OF THALASSEMIA
The major pathophysiology of thalassemia is an imbalance in the ratio of globin chains, which is normally well controlled. Unbound globin chains (i.e., α-globin in β-thalassemia and β-globin in α-thalassemia) precipitate, leading to the destruction of erythroid precursors. RBC precursor damage leads to ineffective erythropoiesis in the bone marrow, and RBC hemolysis in circulation [8, 9].
α-thalassemia
The major cause of α-thalassemia is deletions involving one or more α-globin genes with variable lengths of the α-globin locus, which account for approximately 95% of α-thalassemia cases [10]. α-globin genes are duplicated and localized into two highly homologous units, and unequal crossover between these units during meiosis is likely to be the underlying mechanism of gene deletion [1]. Normal individuals have two α-globin genes on each chromosome. In diagnostic practice, α-thalassemia is classified into 1) α0-thalassemia, in which both α-globin genes are deleted (--/), and 2) α+-thalassemia, in which one of two α-globin genes is deleted (-α/) [11]. Less frequently, α+-thalassemia results from non-deletional variants [i.e., single nucleotide variants (SNV) or short insertion/deletions] [αTα/(α2-globin gene is affected) or ααT/(α1-globin gene is affected)] (Table 1).
In α0-thalassemia, the two most common deletion forms are (/--SEA, South Asia) and (/--MED, Mediterranean). Another type of rare deletion leading to α0-thalassemia is the deletion of the MCS, in which the α-globin genes remain intact, but are completely inactivated [5, 11]. In α-thalassemia, -α3.7 and -α4.2 are the most common forms. Unequal recombination between two homologous segments (Z boxes) that are 3.7 kb apart results in the formation of a chromosome with one α-globin gene (/-α3.7), and similarly, the other mispaired homologous segment (X boxes), 4.2 kb apart, produces the /-α4.2 allele [12]. The non-deletional type includes SNVs or short insertion/deletions in the α-globin gene, or in regions that affect α-globin expression. More than two-thirds of these variants are observed in the α2-globin gene, while less than one-third of the variants are observed in the α1-globin gene. The products of α2-globin genes account for the majority (∼two-thirds) of the total α-globin, while the α1-globin gene accounts for the remainder. Therefore, non-deletional variants in the α2-globin gene would elicit more severe effects than non-deletional variants in the α1-globin gene [13]. In addition, non-deletional variants lead to a greater reduction in α-globin chain expression than the single α-globin gene deletion form of thalassemia [12, 13]. The most common forms that occur in the α2-globin gene are αIVS1(-5nt)α (Mediterranean, 5 nucleotide deletion in IVS1), αPA(AATAAG)α (Middle East Asia, 3′ untranslated region [UTR] polyadenylation site variant), and αCSα (South Asia, stop codon variant resulting in protein extension by an additional 32 amino acids) [14]. Table 2 presents the distribution of the α-thalassemia genotypes identified in the Seoul National University Hospital, South Korea.
β-thalassemia
Unlike α-thalassemia, which is primarily caused by deletions, the majority of β-thalassemia-causing variants are non-deletions, including single nucleotide substitutions and short insertion/deletions leading to frameshift [15, 16]. This disorder is heterogeneous at the molecular level, and more than 300 variants of the β-globin gene have been identified thus far [16]. According to the degree of quantitative reduction in the production of β-globin, β-thalassemia alleles are classified into three categories: 1) the absence of β-globin (β0); 2) β-globin is produced but reduced (β+); and 3) β-globin production is minimally reduced (β++, also known as silent).
The categories of non-deletional variants of β-thalassemia are presented in Table 3. Non-deletional variants in β-thalassemia affect one of the following steps: transcription, RNA processing, or translation. Transcriptional variants involve promoter regions or the 5′ UTR. This category of variants generally results in mild, and occasionally silent, reduction of β-globin synthesis (β+ and β++). Variants that interfere with primary RNA transcript processing are usually found on either canonical or cryptic splice sites, and can lead to either β0, β+, or β++ thalassemia, depending on the proportion of abnormal mRNA transcripts produced. Other variants that reduce the efficiency of RNA processing include polyadenylation (poly-A) signal variants and those in the 3′ UTR, which generally cause β+ thalassemia [15, 17]. Variants that produce a premature termination codon (e.g., nonsense or frameshift) account for the most common types of β-thalassemia, and lead to β0 thalassemia. However, truncating variants in the last exon (exon 3) and the 3′ half of exon 2 are predicted to escape nonsense-mediated decay and produce truncated β-globin to form hemoglobin tetramers that are highly unstable and non-functional, with a dominant negative effect [16]. Variants involving the initiation codon (ATG) leading to β0 thalassemia have also been identified. In rare cases of β-thalassemia, deletions have been reported and classified into three categories: 1) deletions restricted to the β-globin gene from 105 bp to 67 kb in size; 2) deletions of β-LCR, leaving the β-globin gene intact, yet inactivated; and 3) deletions of β-LCR, which removes most of the β-globin gene cluster, including the β-globin gene [15, 16, 18]. Table 4 presents the molecular spectrum of β-thalassemia identified at the Seoul National University Hospital, South Korea.
CURRENT MOLECULAR DIAGNOSIS
A number of molecular techniques for detecting globin gene variants have been developed. Different strategies should be applied to each variant type, which can be divided into two groups: 1) non-deletional variants, including single nucleotide substitutions and short insertion/deletions, and 2) large deletions and duplications. Disease-causing variants in thalassemia are often population specific, and each population has frequently detected thalassemia alleles [19-21]. Occasionally, the clinical manifestations of thalassemia depend on the type of variant and its location within the gene. For example, in α-thalassemia, non-deletional variants of α-globin genes are associated with more severe phenotypes compared to large deletions [7]. Thus, strategic selection according to the type of variant associated with a specific population and clinical phenotype need to be made in a diagnostic laboratory.
The following discussion focuses on the molecular techniques available in a clinical laboratory, depending on the type of variant and prior knowledge of the variant to be examined (Table 5).
Deletions
Gap-polymerase chain reaction can be applied to common deletions in a specific population, using primers flanking known breakpoints. Common single α-globin-gene deletions include a 3.7 kb deletion (-α3.7) and a 4.2 kb deletion (-α4.2). Moreover, two common α-globin-gene deletions include founder variants in specific populations, such as --SEA (Southeast Asian), --FIL (Filipino), and --MED (Mediterranean). The multiplex ligation-dependent probe amplification (MLPA) method is another technique for characterizing deletions in thalassemia, which can detect both known and unknown deletions. MLPA uses two separate oligonucleotide probes (left probe oligonucleotide and right probe oligonucleotide) that are hybridized to adjacent target sequences and ligated. Ligated probes are amplified via PCR, and the amount of amplified probe ligation products enables the quantification of gene copy numbers. MLPA is simple to perform in clinical laboratories and is suitable for the detection of various deletions [4, 22].
Non-deletions
Several cost-effective methods, such as allele-specific PCR, reverse dot blotting, denaturing gradient gel electrophoresis, and the amplification refractory mutation system can be applied to detect common sequence variants (e.g., Hb Constant Spring in South Asia). Sanger sequencing (i.e., direct DNA sequencing) is currently the most practical method to comprehensively detect all variants without prior knowledge of variants [22]. During Sanger sequencing, the PCR product is obtained and subsequently sequenced using the Sanger dideoxy termination method [4]. However, sequencing of α-globin genes is complex, as two α-globin genes (HBA1 and HBA2) are almost identical, with a length >1 kb. Moreover, sequences of α-globin genes are more guanine-cytosine-rich than those of the β-globin gene; the optimization of PCR conditions is necessary in clinical applications [22]. Recent advances in next-generation sequencing have enabled the detection of novel and structural variants by targeting specific genes or whole genomes [1].
CONCLUSION
Thalassemia is one of the most commonly inherited Hb disorders. The genetic basis of α- and β-thalassemia and molecular techniques applicable in a clinical laboratory for the diagnosis of thalassemia have been described. Understanding the genetic basis of thalassemia and these molecular techniques will have a strong impact on the accurate molecular diagnosis of thalassemia.
 XML Download
XML Download