

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Myelodysplastic syndrome (MDS) is a collection of clonal hematopoietic disorders characterized by peripheral cytopenia, ineffective hematopoiesis, and an increased risk of progression to acute myeloid leukemia (AML) [1]. It is an increasingly prevalent disease, predominantly affecting patients of advanced age. The median age of onset is the 7th decade, while it is rare in children and young adults. Childhood MDS has been historically categorized as de novo or primary MDS and secondary MDS, defined as MDS from inherited bone marrow failure (BMF) syndromes, acquired BMF, and therapy-related MDS associated with previous cytotoxic therapy [2]. Based on recent advances in the defining mechanisms of leukemogenesis and the role of clonal hematopoiesis as a predisposing factor for myeloid malignancies, there is increasing evidence indicating a potential underlying predisposition syndrome in MDS that was previously considered de novo or primary. Recently, there has been a growing awareness of non-syndromic familial MDS predisposition syndromes, including those caused by gene mutations such as ANKRD6, GATA2, ETV6, RUNX1, etc. [3]. Recognizing patients with potential hereditary syndromes could provide valuable insights for genetic counseling and disease treatment. The increasing awareness of these conditions coupled with efforts to refer for genetic counseling and genetic testing has revealed that as many as 10% of individuals with hematologic malignancies may carry a germline susceptibility [4]. In recognition of the impact of this growing field on clinical care, the World Health Organization, European Leukemia Net, and National Comprehensive Cancer Network have all recently incorporated the consideration of MDS/AML germline predisposition syndromes into the MDS/AML classification and clinical management guidelines, making knowledge of these syndromes now essential for clinicians [5-7].
MDS with genetic predisposition (Table 1)
Familial platelet disorder with predisposition to myeloid malignancy: Familial platelet disorder with predisposition to myeloid malignancy (FPDMM) is an autosomal dominant familial MDS/AML syndrome caused by inherited mutations in the hematopoietic transcription factor RUNX1. It typically presents with mild to moderate thrombocytopenia with normal-sized platelets, functional platelet defects leading to prolonged bleeding, and an increased risk of development to MDS, AML, or T-cell acute lymphoblastic leukemia. Recent data suggest that clonal hematopoiesis can be detected in >80% of asymptomatic FPDMM individuals by age 50, providing future means of disease surveillance [8].
The MDS/AML transformation rate in FPDMM is estimated to be 20–60%, with a high degree of variability within families [9]. Although not definitely established, progression to MDS/AML can be associated with the acquisition of a second mutation in RUNX1 or an acquisition of trisomy 21, consistent with the need for a “second-hit” event to initiate malignant transformation [10, 11].
ANKRD26-related thrombocytopenia: ANKRD26-related thrombocytopenia (ANKRD26-RT) is characterized by lifelong mild-to-moderate thrombocytopenia with a normal platelet size and no syndromic associations. Most individuals have normal hemostasis or a mild bleeding phenotype and do not present with severe spontaneous bleeding. The risk for myeloid malignancies, including MDS, AML, and chronic myelogenous leukemia (CML), is increased in individuals with ANKRD26 pathogenic variants [12].
Bone marrow morphology in affected individuals may demonstrate dysmegakaryopoiesis with hypolobulated micromegakaryocytes at baseline, presenting a diagnostic challenge of appropriately distinguishing individuals with germline ANKRD27 mutations versus dysmegakaryopoiesis related to the development of MDS [13]. Among the roughly 222 reported cases of ANKRD26-RT to date, there has been an increased incidence of myeloid malignancies, with 4.9% of patients developing acute leukemias, 2.2% developing MDS, and 1.3% developing CML, yielding an estimated 23-fold, 12-fold, and 21-fold risk for these malignancies, respectively, than the general population [14].
Familial MDS with mutated GATA2 (GATA2 deficiency): Germline mutation in GATA2 can lead to GATA2 deficiency, characterized by a complex multi-system disorder with various manifestations, including variable cytopenias, bone marrow failure, MDS/AML, and severe immunodeficiency. To date, around 550 cases with germline GATA2 mutations have been reported in the literature. GATA2 deficiency is a highly penetrant disorder with a progressive course that often rapidly necessitates hematopoietic stem cell transplantation (HSCT) [15].
Describing the presenting symptoms of 57 patients with germline GATA2 deficiency from the National Institutes of Health (NIH) cohort, viral infections were seen in 23%, disseminated non-tuberculosis mycobacterial infections in 28%, MDS/AML in 21%, lymphedema in 9%, and invasive fungal infections in 4% [16]. According to a recent analysis of 25 patients with germline GATA2 mutation, MDS-associated mutations were identified in symptomatic GATA2 patients, including those presenting with overt MDS and with hypocellular/aplastic bone marrows without definitive dysplasia [17]. Healthy relatives of probands harboring the same germline GATA2 mutations had essentially normal marrows devoid of MDS-associated mutations. The findings suggest that abnormal clonal hematopoiesis is a common event in symptomatic germline-mutated GATA2 patients with MDS and those with hypocellular marrows without overt morphologic evidence of dysplasia, possibly indicating a pre-MDS stage warranting close monitoring for disease progression.
The management of patients with GATA2 deficiency often involves a multidisciplinary care team due to the multi-organ involvement. The high incidence of MDS/AML in these individuals warrants regular evaluation of the blood to check for signs of worsening immunodeficiency and monitor the blood counts.
DDX41-associated familial MDS/AML syndrome: The DDX41-associated familial MDS/AML syndrome is a recently identified autosomal dominant syndrome presenting in mid to late adulthood. This is caused by germline mutations in the DEAD-Box helicase DDX41, leading to altered pre-mRNA splicing and RNA processing [18]. DDX41 mutations result in an increased lifetime risk of myeloid neoplasms, including MDS, AML and CML. The average age at diagnosis is 60 years, making it difficult to clinically distinguish patients with de novo MDS/AML from those with a germline mutation in DDX41. This complication may result in its underdiagnosis. The prevalence of DDX41 germline mutations is not known. However, in a cohort study of 1385 MDS or AML patients, DDX41 variants were identified in 43 patients (3.1%), and causal variants were seen in 33 patients (2.4%) [19]. These mutations are relatively common in adult MDS/AML, often without a family history, decreasing the need for systemic screening. Other than the significant family history, DDX41-related malignancies have no apparent preceding clinical signs or symptoms related to the increased risk of hematologic malignancy. Similar to other predisposing syndromes, families with known DDX41 mutations require a bone marrow biopsy at diagnosis with cytogenetic analysis and a complete blood count at regular intervals [20].
SRP72-associated MDS: Germline mutations in the ribonucleoprotein complex gene SRP72 have been identified as a rare cause of familial MDS and bone marrow failure. Mutations in SRP72 were detected in two families with aplastic anemia and MDS. In both families, auditory abnormalities were found, and MDS developed in adulthood. Given the rarity of these germline mutations, little is known regarding their incidence and risk for hematologic malignancies [21].
ETV6-associated familial thrombocytopenia and hematologic malignancy: Patients with thrombocytopenia 5 have an autosomal dominant disorder of the thrombocytopenia, leading to increased susceptibility to bleed. Symptoms usually present during childhood, and patients have been found to have germline mutations in ETV6. The common phenotype is mild to moderate thrombocytopenia with a variable predisposition to MDS, AML, and ALL. Recently, Di Paola and Porter [22] reported that thrombocytopenia was almost completely penetrant, and leukemia was reported in around 30% of carriers with the highest incidence of ALL. Early-onset colorectal cancer has also been reported in a small number of individuals [23].
Table 1
Myelodysplastic syndrome with genetic predisposition.
![]()
Detection and management of patients and family members with MDS with genetic predisposition
Any family with a pattern of familial thrombocytopenia and a predisposition to hematological malignancies should be screened for germline mutations. Recognizing individuals with MDS who may harbor germline mutations can be significantly enhanced by obtaining a careful medical and family history. Guidelines for the clinical detection of familial MDS syndromes have been proposed by Churpek et al. [20]. An algorithm for screening newly diagnosed myeloid malignancy patients for further referral to genetic counseling and testing is shown in Fig. 1 [4, 20].
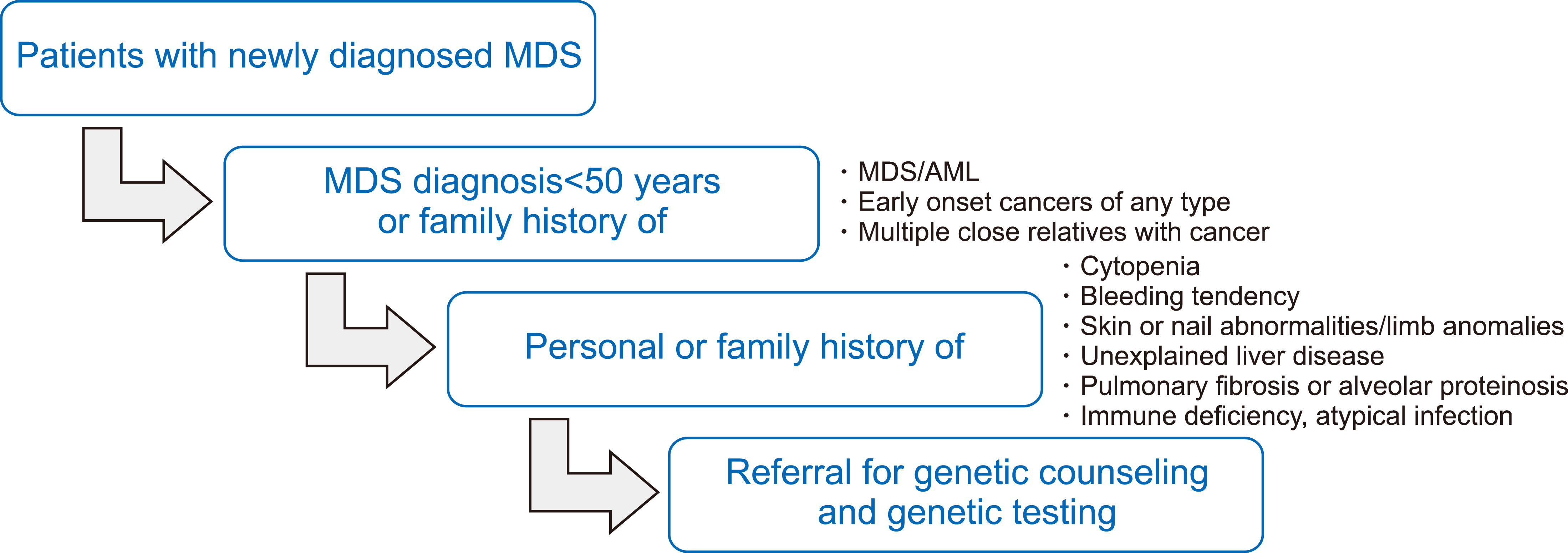
A DNA sample derived from cultured skin fibroblasts remains the gold standard for germline testing. However, if skin fibroblasts are not available, other tissues, such as cultured bone marrow-derived stromal cells, may be used, depending on the laboratory. Also, segregating mutations can be screened if DNA samples are obtained from multiple family members. There is an increasing availability of multigene panel testing for cancer predispositions and platelet disorders. A significant challenge for diagnostic laboratories and clinicians is the classification of variants, which is especially difficult for rare genetic diseases with evolving phenotypes.
Once there is evidence for a pathogenic germline mutation, patients should undergo periodic follow-up with a complete blood count, clinical examination, and a bone marrow biopsy at baseline. A careful clinical examination and regular screening for associated malignancies are recommended, and genetic counseling and site-specific testing should be offered to any family member at risk.
In contrast to sporadic or primary MDS, where the prognostic risk stratification guides the treatment plan, current MDS prognostic models have excluded patients with known genetic predisposition syndromes, greatly underestimating the risk of progression in most cases [24]. The long-term outcomes of MDS secondary to familial MDS/AML predisposition are historically poor, driven by progressive cytopenia, infectious complications, and evolution to acute leukemia. For these patients, HSCT should be considered earlier in the disease course [25, 26]. Any consideration for cytotoxic therapy prior to HSCT must be weighed with the risks of treatment-related toxicity. Further, familial MDS/AML predisposition adds several considerations to the pre-transplant planning and post-HSCT care of an MDS patient, particularly on the choice of a conditioning regimen, selection of HSCT donor, and unique-transplant-related toxicities. Matched-related stem cell donors should be considered very carefully, and donors with known germline mutation or unclear carrier status should be avoided.
Go to : 
CONCLUSIONS
Increasing awareness about germline predisposition and the widespread application of unbiased whole-exome sequencing has contributed to the discovery of new clinical entities with high risks of developing into hematopoietic malignancies with genetic predispositions. The identification and management of individuals with genetic predisposition is a current challenge for hematologists. Importantly, several features such as familial and personal history and molecular and cytogenetic findings may help clinicians consider an underlying genetic predisposition. It is strongly recommended that precise diagnosis, genetic counseling, familial screening, and follow-up programs be provided for patients predisposed to such malignancies.
Go to :
 XML Download
XML Download