

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Ureteral obstruction (UO) that leads to tubular atrophy, interstitial inflammation, and fibrosis is a common cause of renal impairment in children and adults [1,2]. The unilateral ureteral obstruction (UUO) model is the most widely used rodent experimental model of the pathophysiology of acute kidney inflammation and chronic renal fibrosis induced by human UO [3]. UUO induces tubular cell death, interstitial inflammatory cell infiltration, capillary rarefaction, and progressive fibrosis with loss of renal parenchyma. UUO-induced inflammation and consequent fibrosis are strongly related to oxidative stress caused by the over-production of reactive oxygen species (ROS) or downregulation of ROS scavengers [4].
There are various ROS scavengers, including superoxide dismutases, catalases, glutathione peroxidases, and thiol-containing enzymes such as thioredoxins, thioredoxin reductases, peroxiredoxins, and glutaredoxins [5-7]. NAD(P)H plays a critical role in the antioxidant system as a required factor for these antioxidant enzymes [6]. Isocitrate dehydrogenases (IDHs) produce NAD(P)H by reduction of NAD(P) during oxidative decarboxylation of isocitrate to α-ketoglutarate [8]. Among the 3 isotypes of mammal IDHs (IDH1, IDH2, and IDH3), IDH2 is an NADP+-dependent NADPH-producing anti-oxidative enzyme effective against cell dysfunction and cell death and aging such as hair grayness, skin pathology, and renal dysfunction [9]. Accordingly, we have previously reported that IDH2 plays a protective role in various pathologic conditions such as kidney ischemia/reperfusion (I/R) injury [10], liver I/R injury [11], and cisplatin-induced kidney injury through the defense of tissues against oxidative stress [12]. Even though UUO-induced renal injury critically depends on the oxidative stress [4], the role of IDH2 in UUO-induced renal inflammation and the underlying mechanism is largely unknown.
Cells of monocyte/macrophage lineage are often the predominant infiltrating cell types in both experimental models and human renal diseases, and there is a close correlation between the renal infiltration of macrophages and oxidative stress [4,13]. Furthermore, the phenotypes and quantity of infiltrating macrophages are important factors affecting inflammation and fibrosis after UUO [14,15]. Therefore, knowledge of infiltrative macrophages and modulating characteristics of macrophages may yield novel approaches in treating UUO-induced renal inflammation and fibrosis [15]. Here, we investigated the role of IDH2 in UUO-induced oxidative stress, macrophage phenotype change, and infiltration into the kidney, renal inflammation, and the underlying mechanism of action in this setting.
METHODS
Animal experiments
All animal experiments were conducted in accordance with the guidelines provided by the Animal Care and Use Committee of Kyungpook National University. The experimental protocol (KNU 2014-0058) was approved by the Institutional Animal Care and Use Committee at Kyungpook National University. Eight- to 10-week-old female IDH2 gene-deleted (IDH2–/–) mice and wild type (IDH2+/+) littermates were used in the experiments. Pan et al. [16] reported that IDH2–/– females are more susceptible to fructose-induced non-alcoholic fatty liver disease and the associated inflammatory response. Therefore, we used female mice because UUO-induced damage largely depends on inflammatory responses. Generation and characterization of IDH2–/– mice have been described previously [17]. Mice were allowed free access to water and standard chow. Mice were anesthetized by intraperitoneal injection of pentobarbital sodium (50 mg/kg body weight; Sigma-Aldrich Corp., St. Louis, MO, USA) before surgery. For UUO surgery, the right kidney was exposed via a flank incision, the right ureter was completely obstructed near the renal pelvis by 6–0 nylon, and the incision was subsequently sutured. Sham operations were conducted according to the same surgical procedure except for the ureter ligation. Kidneys were harvested 5 days after UUO and were either snap-frozen in liquid nitrogen for biochemical analysis or perfusion-fixed in PLP (4% paraformaldehyde, 75 mM L-lysine, and 10 mM sodium periodate; Sigma-Aldrich Corp.) for histological studies [18].
Western blot analysis
Protein samples were prepared by lysis of kidneys or cells with lysis buffer containing 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM Na2 EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 µg/ml leupeptin, 50 µM PMSF, 10 µM pepstatin A, 100 µM benzamidine, and 50 µM bestatin. The lysate was centrifuged at 15,000 rpm at 4°C. The supernatant was taken and the concentrations of proteins were measured using a BCA kit (Thermo Fisher Scientific, San Diego, CA, USA). Lysates and 5X SDS-PAGE loading buffer (Intron, Seongnam, Korea) were mixed and heated at 98°C for 5 min. SDS samples were stored at –20°C until use. Protein samples were electrophoresed on 10% to 12% polyacrylamide gel with 0.1% SDS and transferred to PVDF membranes, and then subjected to immunoblotting with antibodies against IDH2 [19], 4-hydroxynonenal (HNE; Abcam Inc., Cambridge, MA, USA), and lymphocyte antigen 6 complex (Ly6G; eBioscience, San Diego, CA, USA), For loading control, anti- glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Novus Biologicals, Littleton, CO, USA) antibody was used. Horseradish peroxidase-conjugated secondary antibodies (anti-rabbit: Santa Cruz, Santa Cruz, CA, USA; anti-mouse: Bethyl, Montgomery, TX, USA) were applied. Immunoblots were visualized using chemiluminescence reagent (PerkinElmer Life Sciences, Waltham, MA, USA). Immunoblot densities were quantified using image analysis software ImageJ (NIH, Bethesda, MD, USA).
Isolation of mitochondria from kidney tissue
The procedures for the fraction is as follows [20]. Briefly, kidney tissue was homogenized in sucrose buffer (0.2 M sucrose, 1 mM EGTA, 10 mM HEPES, pH 7.4; Sigma-Aldrich Corp.) using a Teflon homogenizer (Daihan Scientific, Seoul, Korea). The homogenate was centrifuged at 600 × g for 10 min at 4°C. The supernatant was transferred to a new tube and centrifuged at 7,000 × g for 10 min. The mitochondrial pellet was washed twice with sucrose buffer and centrifuged at 7,000 × g for 10 min. The pellet was suspended with sonication buffer (0.1% Triton X-100 in PBS) and sonicated twice using a 4710 series sonicator (Cole-Palmer, Chicago, IL, USA) at a 40% output setting for 10 sec. After centrifugation at 15,000 × g for 30 min, the supernatant containing mitochondria was collected. It was used to measure the mitochondrial hydrogen peroxide levels and oxidative stress.
Measurement of mitochondrial hydrogen peroxide levels in the kidney
Xylenol orange (Sigma-Aldrich Corp.), a ferric-sensitive dye, was used to measure the hydrogen peroxide levels in the kidney mitochondrial homogenates. This measurement is based on the following principle: H2O2 oxidizes iron (II) to iron (III) in the presence of sorbitol, which acts as a catalyst. Iron (III) makes a purple complex with xylenol orange. The procedure is as follows. 100 µg kidney mitochondrial homogenates were incubated with 900 µl Fox reagent containing 0.25 M H2SO4, 1 M sorbitol, 25 mM ferrous ammonium sulfate, 1 mM xylenol orange at room temperature for 30 min. Then, the absorbance was read at 560 nm using a microplate reader (GEMINI EM, Molecular Devices, Sunnyvale, CA, USA). Using this value, hydrogen peroxide levels were calculated.
Immunohistochemistry staining
PLP-fixed kidneys were washed with PBS three times for 5 min each time, embedded in paraffin, and 3 µm thick-sectioned using a microtome (RM2165; Leica, Bensheim, Germany). Immunohistochemical staining was performed as described previously using anti-F4/80 antibody (Serotec, Oxford, UK). Briefly, deparaffinization and rehydration were performed by successive immersion for 10 min in xylene, ethanol (100%, 90%, 80%, and 70%; v/v), and distilled water. After a 5-min permeabilization period with 0.1% Triton X-100/PBS (w/v) and a 1 h blocking with 3% BSA/PBS (w/v), the sections were incubated with primary antibody overnight at 4°C. The sections were washed three times in PBS for 5 min each, incubated with HRP-conjugated secondary antibody (anti-rat IgG; Bethyl Laboratories). Then sections were incubated with DAB substrate (Vector Laboratories, Inc., Burlingame, CA, USA). Hematoxylin and eosin (H&E) staining (Muto Pure Chemicals, Tokyo, Japan) was used to visualize nuclei. Images were captured using a Leica DM2500 microscope (Leica instruments, Wezlar, Germany). F4/80-positive cells were counted in 10 fields (mm2 per field) per kidney.
Bone marrow-derived macrophage preparation
Bone marrow was collected from femurs and tibias by flushing with DMEM (Corning, Corning, NY, USA). The collection was filtered through a 40 µm pore cell strainer and then RBCs were lysed using RBC lysis buffer. Cells were washed with PBS and incubated on the culture dish for 4 h at 37°C. Supernatants were collected and centrifuged. The pellet was resuspended and then incubated in DMEM supplemented with 10% FBS (Welgene Inc., Daegu, Korea), 100 µg/ml streptomycin (Welgene Inc.), 100 U/ml penicillin (1XS/P) (Welgene Inc.), and 20% conditioned medium of L929 cells, which is a source of macrophage colony-stimulating factor (M-CSF), at 37°C for 7 days. Images of macrophages were captured using a Leica DM2500 microscope (Leica instruments, Wezlar, Germany). L929 cell-conditioned medium was obtained from L929 cell culture. L929 cells (ATCC, Manassas, VA, USA) were grown to confluence in DMEM, 10% FBS, and 1X S/P at 37°C. After 3 days, supernatants were collected and used.
Migration assay
Migration of bone marrow-derived macrophages was determined as described previously using a Boyden chamber (Corning Costar, Cambridge, MA, USA) that contains a polycarbonate transwell membrane filter (6.5 mm diameter, 8 µm pore size) with a little modification [21]. 20 × 104 cells were seeded into the insert chamber in DMEM (Corning) with 1% FBS (Gibco, Grand Island, NY, USA). The lower chamber was filled with DMEM with 10% FBS. Cells were incubated for 4 h at 37°C in a humidified atmosphere containing 5% CO2. Cells were migrated through the pores of the membrane at the bottom of the insert chamber. After the remaining cells were scraped from the upper surface of the membrane with a cotton swab, migrated cells on the lower surface of the membrane were stained with hematoxylin and eosin and then counted. Pictures were taken using a microscope (Leica; Leica instruments, Wetzlar, Germany).
Immunofluorescence staining
Bone marrow-derived macrophages cultured on the coverslips were fixed with 4% paraformaldehyde for 15 min, permeabilized with 0.1% Triton-X 100 for 1 min, quenched with 50 mM ammonium chloride for 5 min and then incubated with 3% BSA to block non-specific binding. To achieve dual immunofluorescence staining of Rac1 and IDH2, mouse anti-Rac1 antibody (BD Biosciences, San Jose, CA, USA) and rabbit anti-IDH2 antibody [22] were mixed and used. The slides were incubated with an antibody mixture overnight at 4°C. Slides were washed with PBS, and then Cy3-conjugated anti-mouse IgG (Bethyl Laboratories Inc.) and FITC-conjugated anti-rabbit IgG (Bethyl Laboratories Inc.) were applied for 60 min at room temperature. To detect the cell nuclei, 40-6-diamidino-2-phenylindole (DAPI, #D9542; Sigma Aldrich) was applied. Finally, the cells were observed and pictures were taken using a fluorescence microscope (Leica; Leica instruments, Wetzlar, Germany).
RESULTS
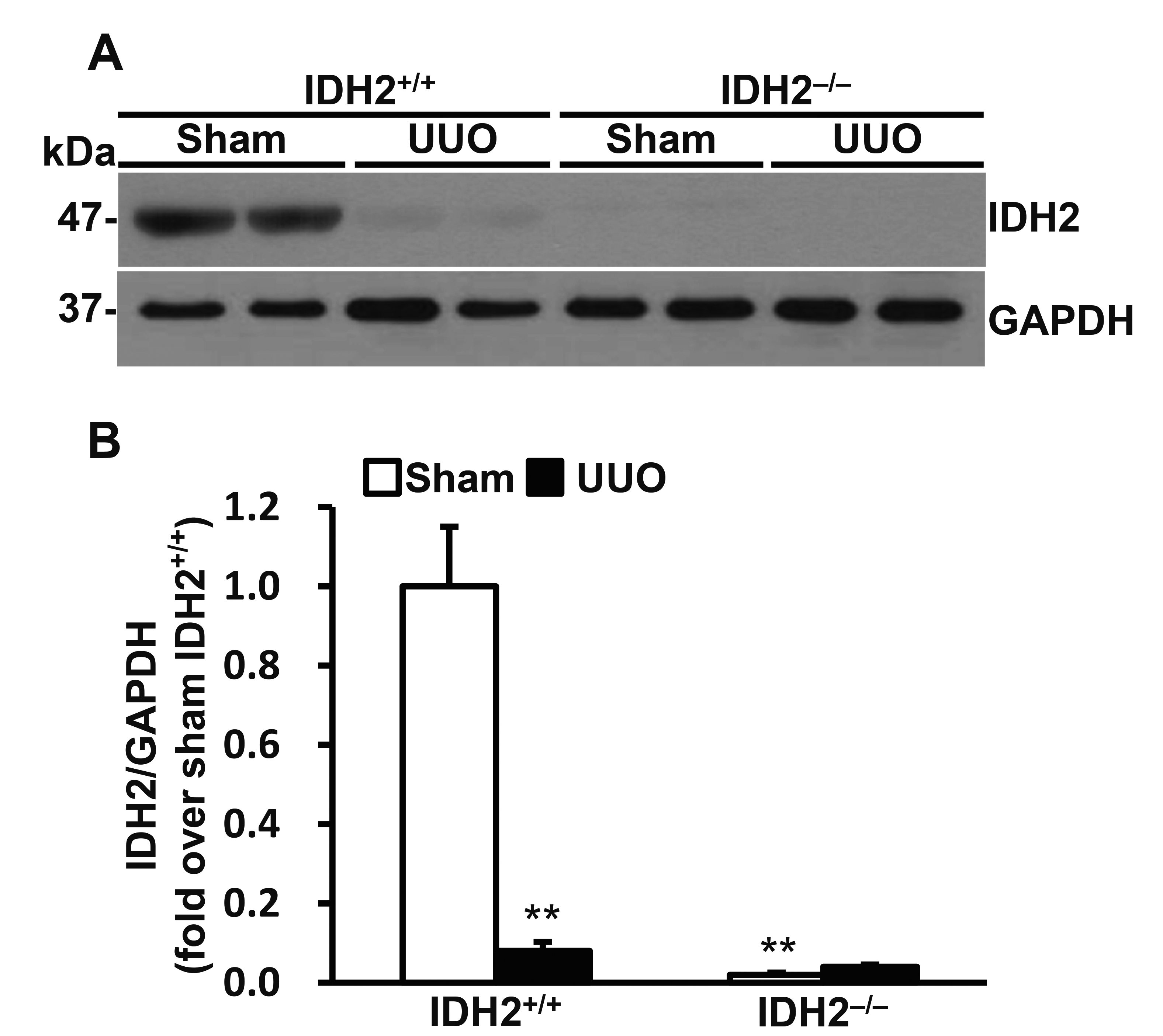
UUO decreased IDH2 expression
To speculate the role of IDH2 in UUO-induced kidney injury we determined the expression levels of IDH2 following UUO. First of all, we confirmed that IDH2 was not expressed in the kidney of IDH2−/− mice. UUO significantly decreased IDH2 expression (p < 0.01 vs. sham) (Fig. 1A, B).
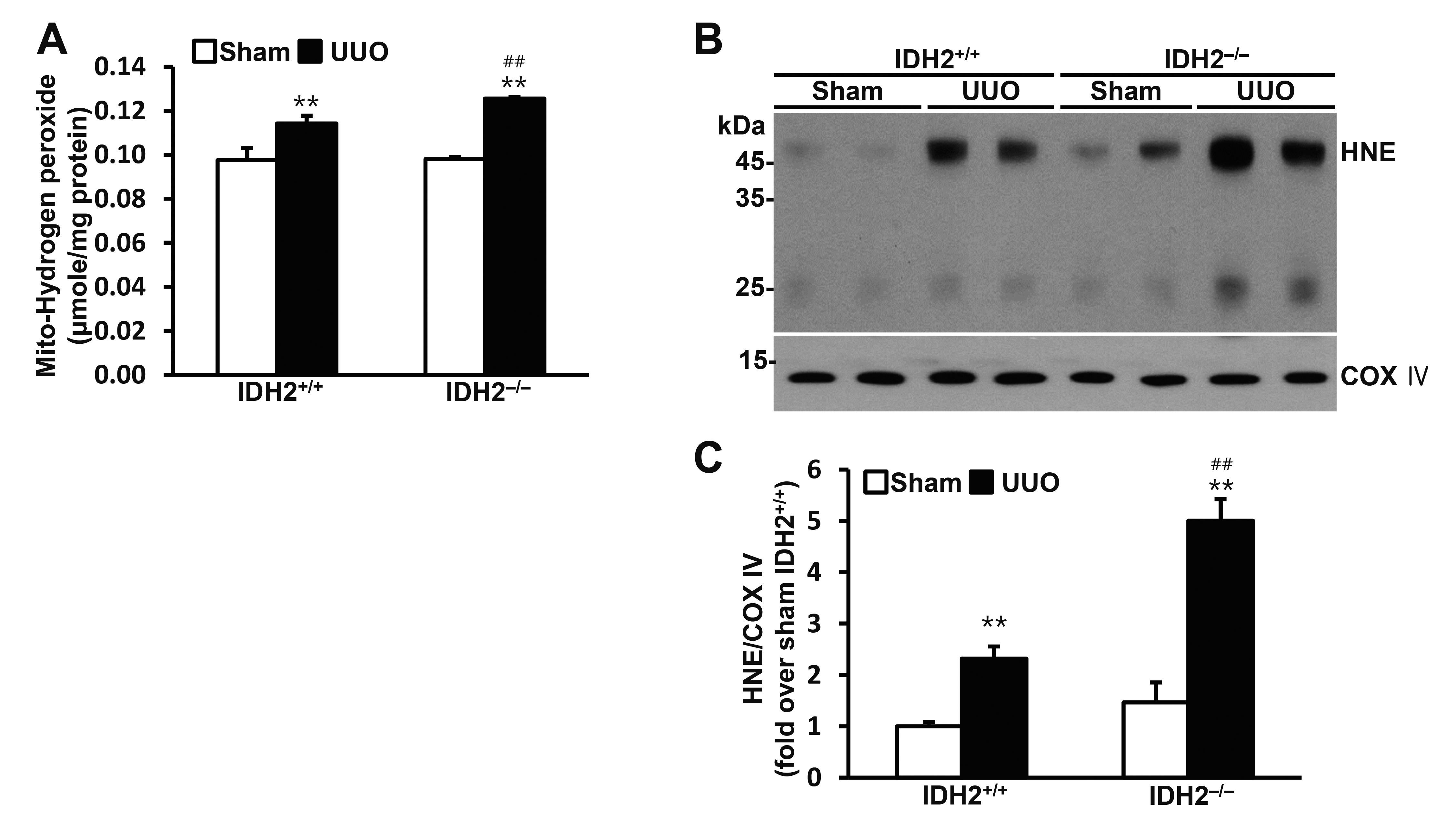
IDH2 gene deficiency accelerated UUO-induced mitochondrial oxidative stress
To investigate the role of IDH2 in mitochondrial oxidative stress, we determined the hydrogen peroxide production and the expression of HNE, which is produced by lipid peroxidation, in the mitochondrial fraction of the kidney cells. Hydrogen peroxide production in the mitochondria was increased by UUO in both IDH2+/+ mice and IDH2−/− mice (p < 0.01 vs. their own sham group), while IDH2 gene deficiency accelerated the UUO-induced increase in hydrogen peroxide production (p < 0.01 IDH2+/+ UUO vs. IDH2−/− UUO) (Fig. 2A). HNE level was also increased by UUO in both IDH2+/+ mice and IDH2−/− mice (p < 0.01 vs. their own sham group), while IDH2 KO accelerated the UUO-induced increase in HNE (p < 0.01 IDH2+/+ UUO vs. IDH2−/− UUO) (Fig. 2B, C). HNE, 4-hydroxynonenal.
IDH2 gene deficiency accelerated UUO-induced inflammatory cell infiltration into the kidney
To investigate the role of IDH2 in UUO-induced inflammation, we determined inflammatory cell infiltration in the kidney following UUO. Expression of Ly6G, which is a marker for neutrophils and monocytes, increased in both IDH2+/+ mice and IDH2−/− mice (p < 0.01 vs. their own sham group), while IDH2 gene deficiency accelerated increase in Ly6G expression in IDH2−/− mice (p < 0.01 IDH2+/+ UUO vs. IDH2−/− UUO) (Fig. 3A, B). Expression of F4/80, which is a macrophage marker, also increased in both IDH2+/+ mice and IDH2−/− mice (p < 0.01 vs. their own sham group), while IDH2 gene deficiency accelerated increase in F4/80 expression in IDH2−/− mice (p < 0.01 IDH2+/+ UUO vs. IDH2−/− UUO) (Fig. 3C, D).
IDH2 gene deficiency accelerated the migration of bone marrow-derived macrophages
To elucidate the mechanism underlying the increased infiltration of inflammatory cells into the kidney, we investigated the effect of IDH2 gene deficiency on the phenotype change of bone marrow-derived macrophages. First, we observed the morphology of the IDH2−/− mice under the microscope. Ruffle formation of the macrophages was enhanced in IDH2−/− mice (Fig. 4A). Next, we determined migration using Boyden chamber. The number of migrated macrophages was greater in IDH2−/− mice compared to IDH2+/+ mice (p < 0.01 IDH2+/+ vs. IDH2−/−) (Fig. 4B, C). Dual immunofluorescence staining against IDH2 and Rac1 showed enhanced ruffle formation with Rac1 spread in IDH2−/− mice compared to IDH2+/+ mice (Fig. 4D).
DISCUSSION
The principal findings of this study are that the NADPH-producing anti-oxidant enzyme IDH2 deficiency increased UUO-induced kidney inflammation through enhanced mitochondrial oxidative stress, phenotype change of macrophages, and infiltration of macrophages into the kidney. UUO also decreased IDH2 expression in IDH2+/+ mice with oxidative stress and inflammation in the kidney indicating that there is a vicious cycle between kidney injury and a decrease in IDH2 expression.
Ureteral obstruction causes tubular cell injury and death with increased macrophage infiltration, the elevation of pro-inflammatory cytokines, and proliferation of interstitial fibroblasts resulting in kidney fibrosis, which is a common pathway to the end-stage of renal disease. Findings in the animal models are comparable with observations in patients with obstructive nephropathy [23] suggesting that the rodent model of UUO is useful to study human progressive renal disease offering the beneficial effects of genetic modulation. Oxidative stress contributes importantly to the pathogenesis of UUO. Various markers of oxidative stress, such as 4-HNE are increased in UUO kidneys [24,25]. In accordance with, mice that are genetically deficient in catalase are more susceptible to UUO-induced renal injury [26]. Our group previously revealed the protective role of IDH2 in I/R–induced kidney injury and cisplatin-induced nephrotoxicity using IDH2−/− mice. In these studies, we demonstrated the anti-inflammatory role and mechanism of anti-mitochondrial oxidative stress and anti-apoptosis by the IDH2 gene [10,12]. However, the role of IDH2 in obstructive kidney disease and the link between IDH2 deficiency and increased inflammation was mostly veiled. Here, we demonstrated that UUO decreased IDH2 and IDH2 deficiency aggravated UUO-induced mitochondrial oxidative stress, macrophage activation, and migration resulting in enhanced renal inflammation for the first time.
With the contribution of ROS by other cells, invading immune cells are the major source of oxidative stress during kidney injury. Infiltrating leukocytes (neutrophils and macrophages) release large amounts of inflammatory cytokines and ROS [27]. Macrophage infiltration depends on phenotypes [15]. For example, mice that had received a bone marrow transplant from angiotensin II type 1 receptor null (Agtr1–/–) mice showed reduced macrophage infiltration after UUO [28]. In our study, bone marrow-derived macrophages, which lack IDH2 showed a more migrating phenotype with enhanced ruffle formation and Rac1 diffusion in the cytosol. IDH2 gene-deficient macrophages migrated more than wild type macrophages. Accordingly, IDH2 gene deficiency increased macrophage infiltration into the kidney after UUO, increasing renal inflammation. Because infiltrating macrophages are major contributors for kidney inflammation [27] and global knockout of IDH2 in this study showed migrating phenotype and increased migration of macrophages, it is suggested that macrophage-selective IDH2 deficiency will give similar results with this study.
It can be expected that the basal ROS level is increased with IDH2 deficiency. However, we could not observe an increased basal ROS level without UUO. It seems that the increase in basal ROS with IDH2 deficiency is minimal. Besides, even UUO-induced increase in hydrogen peroxide level was just 14% in wild type, which suggests that the increase may be undetectable in this system. Nevertheless, it is clear that UUO-induced ROS production and consequent oxidative stress is exaggerated by IDH2 deficiency.
In vitro study of macrophage culture (Fig. 4) showed a more migrating phenotype of IDH2 deficient macrophages without any stimulation but macrophage colony-stimulating factor. Morphology and migration of cells largely depend on cytoskeleton particularly polymerized actin. The effect of ROS on actin polymerization is controversial. Reduction of ROS by free radical scavenger reduced F-actin contents resulting in disassembly of the actin cytoskeleton and reduced neuronal growth cone formation [29]. On the other hand, the NADPH-dependent physiological level of ROS attenuated actin polymerization via actin glutathionylation in the neutrophils [30]. Moreover, mitochondrial ROS levels of IDH2+/+ and IDH2−/− mice macrophages in this study were not significantly different. Therefore, it is not likely that ROS is responsible for the differences between morphology and migration activities of IDH2+/+ and IDH2−/− mice macrophages. We suggest that the differences occur in response to macrophage colony-stimulating factor during macrophage differentiation.
Collectively, our results demonstrate that UUO decreases IDH2 in the kidney, and deficiency of the IDH2 gene increases obstruction-induced oxidative stress to increase inflammation, at least in part, via activation of macrophages. The present study demonstrates the importance of the presence of IDH2 to inhibit UUO-induced oxidative stress and inflammation in the kidney, offering IDH2 as a novel therapeutic target for obstructive renal injury and macrophage activation.


 XML Download
XML Download