

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
The selective serotonin reuptake inhibitor FLX ((7)-N-methyl-3-phenyl-3-(α,α,α-trifluoro-p-tolyloxy)propylamine hydrochloride) is the active molecule in Prozac, shown to be effective in the treatment of depression [1-3]. Besides, FLX has off-target effects as it inhibits the activity of the voltage-dependent Na+ and K+ and Ca2+ channels at the plasma membrane [4,5] and is also distributed inside the cell, in the mitochondria (60–70%), synaptosomes, and other cellular organelles [6].
Cyclic adenosine 3’5’monophosphate (cAMP) and Ca2+ are two major intracellular second messengers of hormones, neuromediators, or cytokines functions. cAMP regulates many cellular responses, including cell growth and differentiation, gene transcription, and protein expression [7]. It is synthesized from ATP via the catalysis by adenylate cyclases (AC) and is inactivated by its hydrolysis into AMP catalyzed by nucleotide phosphodiesterases (PDE) [8]. It has been shown that the cAMP signaling pathway mediates many physiological processes like growth, reproduction and apoptosis [9]. The concentration of cytoplasmic calcium ion ([Ca2+]cyt) also has a major role in cell proliferation, migration, gene regulation, or apoptosis [10,11]. These changes can happen through extracellular Ca2+ entry, or intracellular Ca2+ stores mobilization [11,12] or to a mitochondrial matrix Ca2+ overload, itself probably caused by mitochondrial membrane permeability alterations [13,14].
In previous studies, we have shown that FLX exerts an inhibitory effect on the cAMP response of mouse Leydig tumor cells (mLTC) to the gonadotropins luteinizing hormone (LH) and chorionic gonadotropin through different mechanisms [15]. In order to ascertain whether it is also the case in ovarian tumor cells, potentially sensitive to follicle-stimulating hormone (FSH), we undertook a similar study with COV434 cells [16]. COV434 cells were chosen because they derive from ovarian cells, initially sensitive to the gonadotropin FSH that is central in reproduction control. However, COV434 cells, maybe because of their tumoral status, appeared to be insensitive to FSH and we chose to stimulate them with forskolin (FSK), a direct activator of adenylate cyclase, to study their cAMP pathway. The main purpose of our study was to study the sensitivity of their cAMP pathway to FLX at low concentrations but we also followed its effects on intracytoplasmic Ca2+ and ATP.
Our data show for the first time that FLX promotes a dose-dependent inhibition of FSK-stimulated cAMP accumulation in COV434 cells, reminiscent to its effect on mLTC cells, in response to LH but at lower concentrations (0.6–10 µM) than in mLTC cells (12.5–100 µM) [15]. Moreover, we also show for the first time in COV434 cells that FLX provokes a dose-dependent increase in [Ca2+]cyt and a dose-dependent decrease in ATP level, with significant decrease on this tumor cell line viability at 5–10 µM FLX concentrations, suggesting its possible use as a complementary tool in granulosa tumor treatments.
Go to : 
METHODS
Chemicals and reagents
All chemicals were purchased from Sigma-Aldrich (Saint Quentin Fallavier, France) unless otherwise noted. pGlosensor–TM-22F cyclic AMP plasmid and CellTiter-Blue Cell viability assay (G8080) were from Promega (Charbonnières-les-Bains, France), XtremeGENE HP DNA transfection reagent was from Roche (Boulogne-Billancourt, France).
Cell culture and plasmids, transfections
The COV434 cells (human ovarian granulosa tumor) were obtained from Sigma (France). Cells were expanded in supplemented DMEM medium (Gibco, Invitrogen, 10% fetal bovine serum, 50 µg/ml gentamicin, 10 units of penicillin/ml, and 10 µg/ml streptomycin). The cells were grown at 37°C and 5% CO2 and were used between their 8th and 35th passage.
COV434 cells (about 100,000 cells per well) on a 96-well Greiner white/clear bottom plate (Dutscher, Brumath, France) were transfected with pGlosensor–TM -22F cyclic AMP plasmid using XtremeGENE HP DNA transfection reagent. Thirty minutes before transfection, DNA (100 ng plasmid per well) and XtremeGENE HP DNA transfection reagent (0.3 µl per well) were mixed with serum-free RPMI medium. This plasmid consists of the firefly luciferase sequence fused to that of the PKA cAMP-binding domain in a way that allows control of its enzymatic activity by cyclic AMP. The plates were then incubated overnight at 37°C under 5% CO2 before use in the assays.
cAMP quantitation
Transfection supernatants were removed and replaced with medium deprived of fetal-calf serum (100 µl) and containing the luciferase substrate luciferin as well as 1 mM isobutyl-methyl-xanthine (IBMX) in order to inhibit endogenous nucleotide PDE activity. The plates were incubated for 2 h or 24 h with FLX at various concentrations in a 10 µl volume. Finally, FSK was added in a 10 µl volume in triplicate wells to reach 10 µM concentration. Kinetics of intracellular oxyluciferin luminescence were then recorded using a Polarstar Optima luminometer (BMG Labtech, Champigny-sur-Marne, France).
COV434 cells viability assessment
COV434 cells were seeded in 96-well plates at 100,000 cells/well. Two days later, the medium was replaced with serum-free medium in the absence (control) or presence of FLX (0–10 µM) for 2 h or 24 h at 37°C before the addition of 20 µl of CellTiter-Blue Reagent (Promega, Madison, WI, USA) to each well. After having incubated, changes in fluorescence were recorded with a Spectra Gemini spectrofluorimeter (Molecular Devices, Sunnyvale, CA, USA) at an excitation wavelength of 560 nm and an emission wavelength of 590 nm. The fluorescent signal from the CellTiter-Blue Reagent is proportional to the number of viable cells.
Intracellular adenosine triphosphate (ATP) concentration measurement
After the incubation of cells with or without FLX, the ATP level in cells was measured using the CellTiter-Glo 2.0 Assay (Promega). Standards were prepared from ATP standard (Promega) using serial dilutions to obtain concentrations of 1 × 10–10, 1 × 10–11, and 1 × 10–12 M. The assay buffer and substrate were equilibrated to room temperature, and the buffer was transferred with the substrate. After 30 min, a 50 µl sample of this solution was added to 100 µl luciferin/luciferase reagent in 96-well white plates, and the content was mixed for 2 min and incubation was continued for 10 min at room temperature. The luminescence was read with an integration time of 1,000 ms using an Ascent Luminoskan Luminometer (Thermo-Fisher Scientific, Illkirch, France) with PBS as a blank.
Intracellular Ca2+ concentration measurement
The variation in cytoplasmic Ca2+ concentration ([Ca2+]cyt) was assessed using the fluorescent Ca2+ indicator Fluo4-AM. COV434 cells were incubated with different FLX concentrations for 2 h before adding Fluo4-AM, and new incubation was carried out for 30 min at 37°C in the dark, before three washings with PBS. Changes in fluorescence were recorded with a Spectra Gemini spectrofluorimeter (Molecular Devices) at an excitation wavelength of 494 nm and an emission wavelength of 516 nm. Intracellular Ca2+ level calculations after the incubation period were based on the given dissociation constant Kd (Ca2+) = 345 nM of Fluo4-AM (Molecular Probes). The [Ca2+]cyt was calculated using the equation [Ca2+]cyt = Kd (F – Fmin) / (Fmax – F). Maximum fluorescence was determined using 50 µM calcium ionophore A23187 in the presence of 2 mM Ca2+, and minimal fluorescence was determined in the absence of Ca2+ and the presence of 2 mM EDTA.
Area under curve (AUC) calculations and statistical analyses
The GraphPad 5.0 (GraphPad Software, San Diego, CA, USA) was used for AUC determinations of individual kinetics. Mean, and SEM values for each triplicate AUCs were determined. The AUC ratios were used to compare cAMP response kinetics. One-way ANOVA with Dunnett’s test was also performed using this package. The level of significance was p < 0.05.
Go to :
RESULTS
Effect of fluoxetine on the intracellular cyclic AMP response to FSK
The effects of FLX on FSK-stimulated intracellular cAMP in COV434 cells were measured at FLX concentrations ranging between 0 and 10 µM. Fig. 1 shows that the FSK-stimulated cAMP-dependent oxiluciferin fluorescence kinetics are significantly decreased by FLX in dose-dependent and time-dependent manner as calculated by the AUC. Almost full inhibition was observed after a 2-h preincubation with 10 µM FLX final concentration (Fig. 1A, B) and the same was observed with only 5 µM FLX after a 24-h preincubation before FSK addition (Fig. 1C, D).
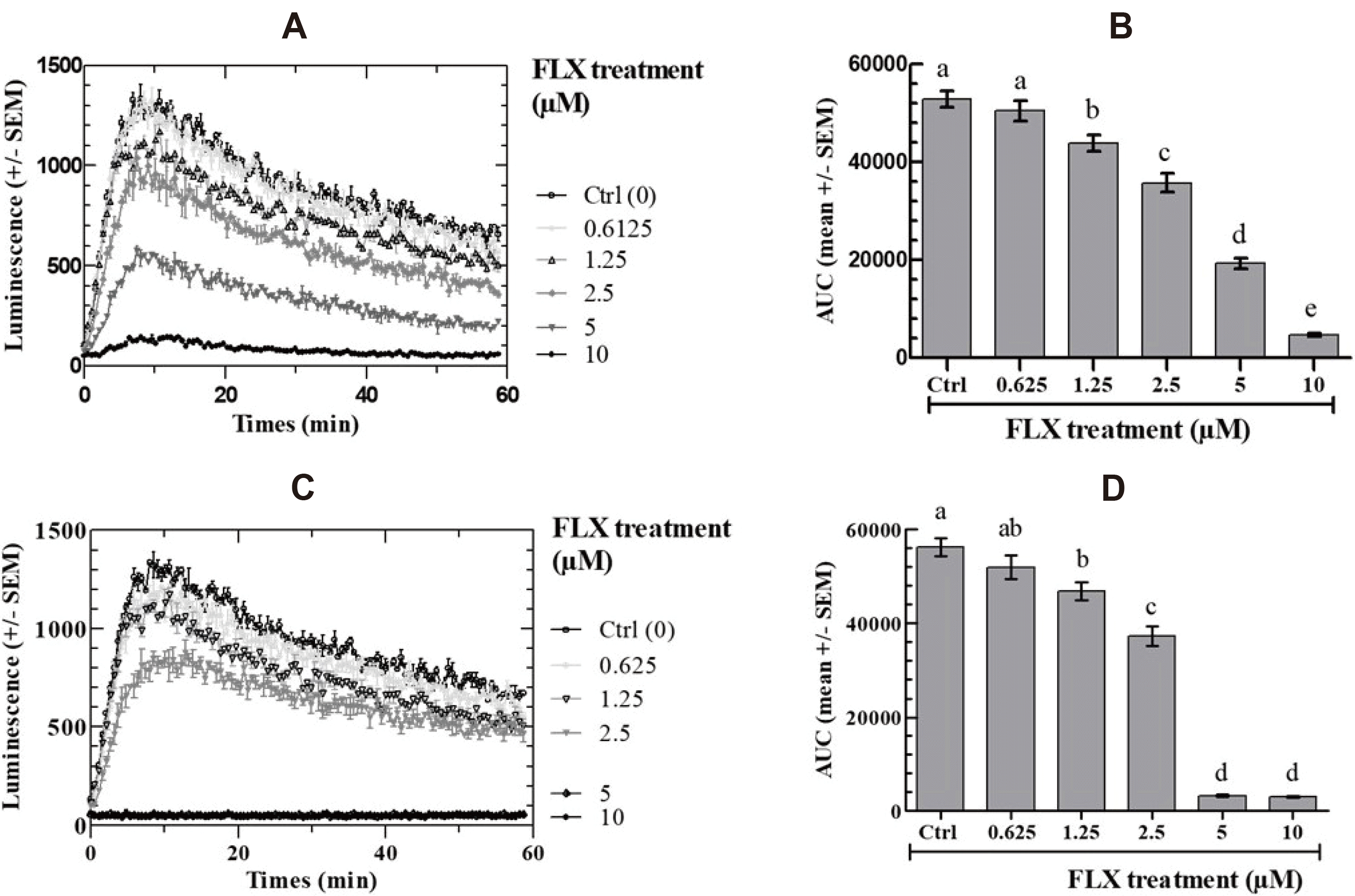 | Fig. 1Effect of fluoxetine (FLX) on intracellular cAMP response of COV434 cells to 10 µM forskolin (FSK).Cells were treated with the indicated concentrations of fluoxetine for 2 h (A, B) or 24 h (C, D). (A, C) Real-time recording of luminescence under stimulation of COV434 cells by 10 µM FSK in the presence of fluoxetine; (B, D) Dose-dependent response to fluoxetine determined by the area under curve (AUC) of individual kinetics in (A, C). Different letters (a–e) in each incubation time indicate significant differences between control and treatment at p < 0.05.
|
Effects of fluoxetine on ATP level in COV434 cells and on their viability
The ATP level in COV434 cells was measured after 2 or 24 h of incubation with various FLX concentrations using the ATP Cell-Titer-Glo Assay. Fig. 2A shows that FLX provokes a dose- and time-dependent decrease in the ATP level.
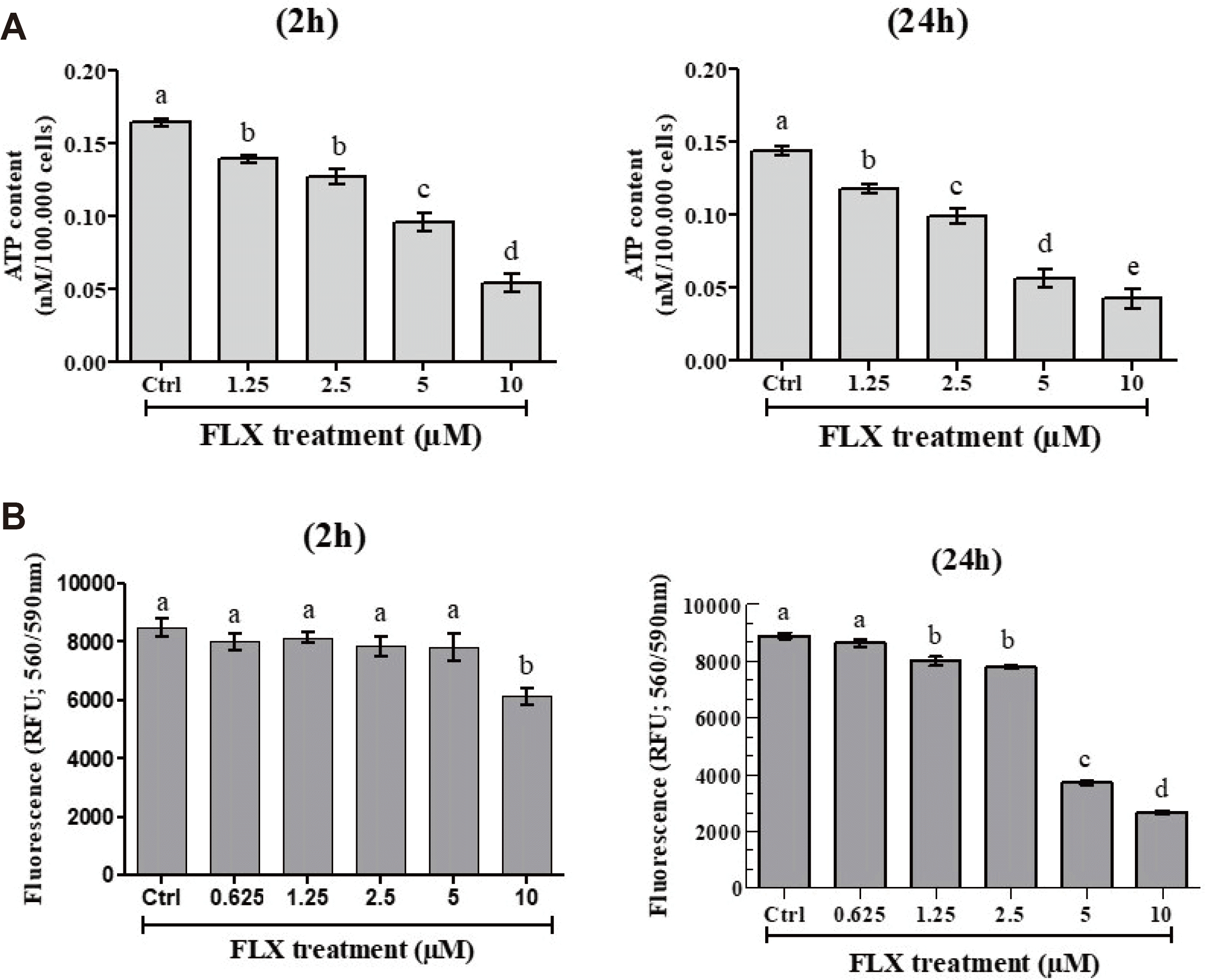 | Fig. 2Effect of fluoxetine (FLX) on ATP levels in COV434 cells and the number of viable COV434 cells.ATP level (A) and the viability of COV434 cells (B) were measured after incubation with various concentrations of FLX for 2 h or 24 h. The experiments were repeated four times; values are mean ± SEM. Different letters (a–e) in each incubation time indicate significant differences between control and treatment at p < 0.05.
|
After 2 or 24 h of incubation in the presence of FLX (0.625–10 µM) and a one-hour further incubation with CellTiter-Blue Assay, cell viability was found to be unaffected when compared to control at all concentrations, except at 10 µM for 2 h and ≥ 5 µM for 24 h (Fig. 2B).
Effects of fluoxetine on cytoplasmic Ca2+ in COV434 cells
The cytoplasmic free Ca2+ concentration ([Ca2+]cyt) in intact cells, calculated before the addition of 5 mM extracellular Ca2+, was approximately 80 nM at 37°C. FLX at concentrations between 0.6125 and 10 µM induced a dose-dependent rapid rise of [Ca2+]cyt up to a plateau at approximately 900 nM (Fig. 3A, B). Thus, the addition of FLX induced a sustained increase in [Ca2+]cyt when Ca2+ is present in the culture medium. In the absence of Ca2+ in the medium, FLX also led to an increase in [Ca2+]cyt, but this increase was only transient (Fig. 3C). When Ca2+ was introduced in the culture medium, [Ca2+]cyt was again increased (Fig. 3D).
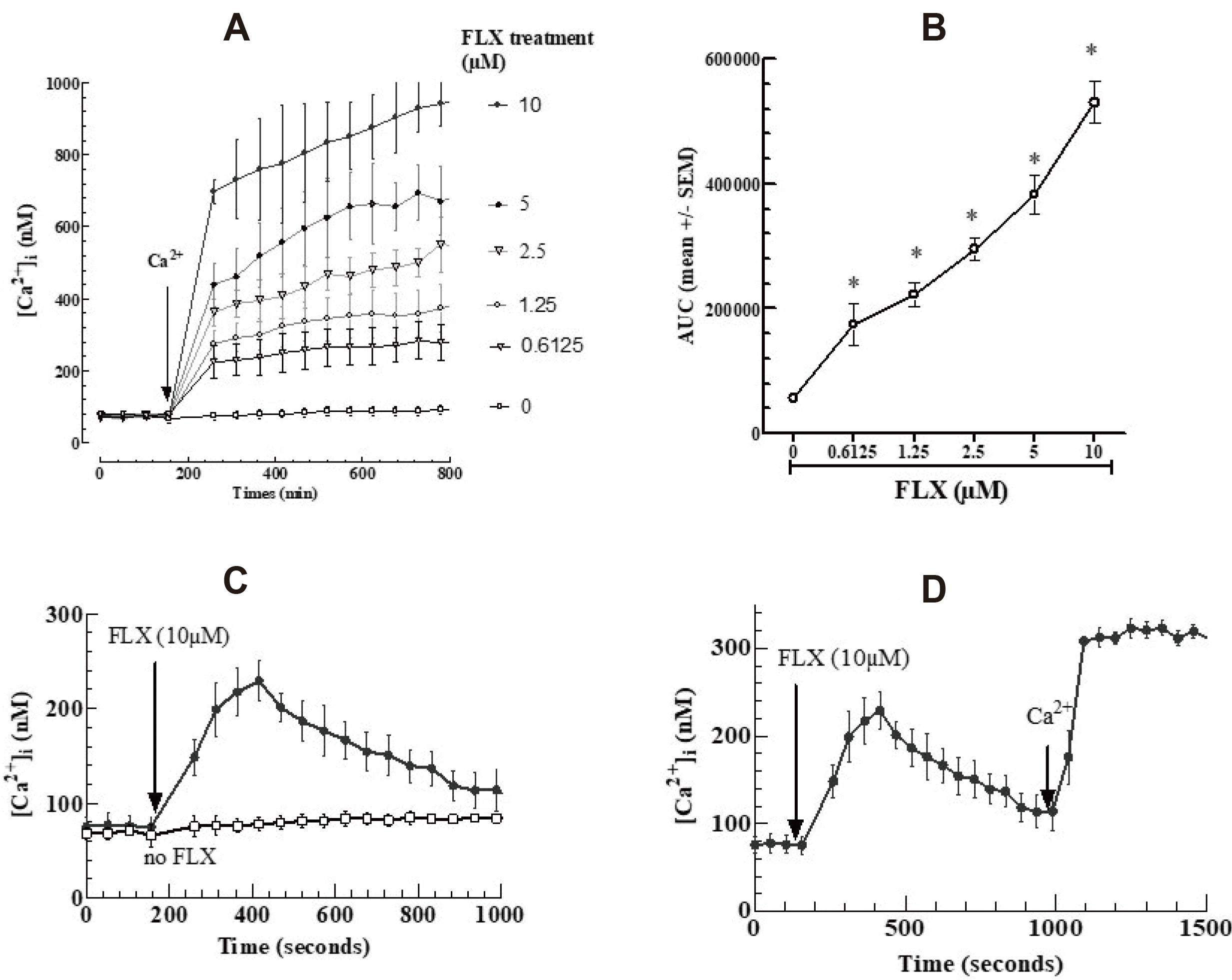 | Fig. 3Effect of fluoxetine (FLX) on [Ca2+]cyt in Fluo4-AM-loaded COV434 cells.Cells were incubated with Fluo4-AM as described in experimental procedures. The intracellular Ca2+ levels were measured spectrofluorimetry using an excitation wavelength of 494 nm and an emission wavelength of 516 nm. The [Ca2+]cyt measurements were made every 52 sec. (A) Baseline levels of [Ca2+]cyt were monitored for 156 sec prior to adding Ca2+ at 2 mM final concentration, changes in [Ca2+]cyt were monitored for an additional 800 sec. (B) Dose-dependent response to fluoxetine determined by the area under curve (AUC) of individual kinetics in (A). (C) Signal of Ca2+ responses induced by FLX (10 µM) in a Ca2+-free medium and Control (Ctrl) (absence of FLX). (D) Cells loaded with Fluo4-AM were incubated first with 10 µM FLX in Ca2+-free medium, and external Ca2+ was added after 1,000 sec. Asterisks in each concentration indicate significant differences between control and treatment at p < 0.05.
|
Go to :
DISCUSSION
We have previously documented that fluoxetine (FLX) exerts a dose-dependent and time-dependent inhibition of LH-stimulated cAMP accumulation in mouse Leydig tumoral cells. The data in the present paper demonstrate that FLX also rapidly inhibits cAMP accumulation under FSK stimulation in human ovarian tumor COV434 cells. After 24 h of incubation, 5 µM FLX almost completely abolished the FSK-stimulated cAMP accumulation response. A final 10 µM FLX concentration was enough to get the same inhibition after only 2 h of incubation. These results are very similar to those observed with FLX on the LH-dependent stimulation of cAMP accumulation in mLTC cells, suggesting a common mechanism.
In order to decipher more precisely the mechanisms of FLX inhibiting cAMP accumulation in COV434 cells, we decided to also study its effects on intracellular ATP and Ca2+ levels as well as on cell survival. In COV434 cells, FLX caused a dose-dependent drop in intracellular ATP concentrations after 2 and 24 h incubations but with only a marginal difference between the two incubation durations. Interestingly, there was no effect of FLX on COV434 cells survival after 2 h except a slight one at the highest dose tested. After 24 h, there was a marked effect of the highest FLX concentrations tested (5–10 µM) on cell survival.
In brief, the above data show that the drop in the cAMP response to FSK as a function of FLX concentration might largely be related to diminished ATP content and but not to diminished cell viability. This is not unexpected as ATP is the substrate for adenylate cyclase to synthesize cAMP. Many previous studies have shown that FLX indirectly affects electron transport and (F1–F0)-ATPase activity, and thus inhibits oxidative phosphorylation in mitochondria, leading to reduce ATP [17]. However, these effects of FLX on mitochondrial activities may result from interacting with VDAC and decreasing its conductance as a channel providing passage for Ca2+ [18], adenine nucleotides [19], other metabolites [20,21], as well as preventing PTP opening by preventing the release of accumulated Ca2+ and by swelling energized mitochondria and inhibiting release of cytochrome C from mitochondria [22].
In order to study more thoroughly the mechanism of the FLX effects on COV434 cells, we have measured the effects of 0.6125 to 10 µM FLX on [Ca2+]cyt. Upon Ca2+ addition in the medium, there was an immediate rise in [Ca2+]cyt that was proportional to FLX concentration in the medium up to a plateau at approximately 900 nM at the highest FLX concentration. In the absence of Ca2+ in the medium, FLX induced an immediate and rapid rise of intracellular [Ca2+]cyt and a secondary [Ca2+]cyt rise was observed upon addition of Ca2+ in the medium. These data indicate that FLX stimulates an increase in intracellular [Ca2+]cyt both from intracellular stores and from outside the cell. Charles et al. [23], have shown that FLX induces an increase in [Ca2+]cyt by emptying the endoplasmic reticulum (ER) through the translocon, an ER Ca2+ leakage structure, that it also inhibits oxygen consumption and lowers mitochondrial ATP, leading to Ca2+ reuptake into the ER, and that Ca2+ quickly accumulates in the mitochondria, leading to mitochondrial Ca2+ overload and cell death [23].
Our data also show that concentration-dependent FLX-induced cell death occurs at the same concentration ranges that induce [Ca2+]cyt rise. Normal cell viability can be altered by either Ca2+-dependent or -independent mechanism [24,25]. Many reports also suggest that 10–100 µM FLX induces cell death in tumor cell types such as colon cancer cells [26], Burkitt’s lymphoma cells [27], and ovarian carcinoma cells [28]. Our data also show that FLX induces cell death in COV434 cells, but at lower concentrations (5–10 µM) than in those cells. This cytotoxicity could be caused by mitochondrial Ca2+ overload, as it has already been shown in glioma cells [29]. At moderate concentration, mitochondrial Ca2+ supports ATP production by stimulating mitochondrial metabolic enzymes such as FAD-glycerol phosphate dehydrogenase, pyruvate dehydrogenase phosphatase, NAD-isocitrate dehydrogenase, oxoglutarate dehydrogenase, F1–F0 ATP synthase, cytochrome C oxidase [30]. But excess Ca2+ in the mitochondria leads to activation of both mitochondria-specific sodium/calcium exchanger (NCLX) and permeability transition pore (PTP) [31,32]. Such prolonged mitochondrial calcium permeability might end up in either apoptosis or necrosis, depending on the availability of ATP since previous works reported that weakly damaged mitochondria still contain enough ATP to trigger apoptosis, whereas more severely damaged mitochondria promote cell necrosis [33-36].
Collectively, the results show that, via direct and/or indirect mechanisms, fluoxetine induced 1) a decreased cAMP response to FSK, 2) a decreased intracellular ATP content, 3) an increased [Ca2+]cyt concentration and 4) a decreased cell viability (only at the highest FLX concentrations tested [5–10 µM], but not at the lowest concentrations [0.6–2.5 µM] already affecting the cAMP, ATP and Ca2+ responses). This last effect favors the use of the anti-depressant drug fluoxetine as a complementary tool, in granulosa tumor treatments, as already proposed for other cancer cell types.
Go to :
 XML Download
XML Download