

 PDF
PDF Citation
Citation Print
Print



서 론
신경섬유종증은 인종, 성별과 관계없이 2,500에서 3,000명당 한 명씩 발생하는 가장 흔한 상염색체 우성 질환 중 하나이다[1]. 신경섬유종증은 제1형(NF1)과 제2형(NF2)으로 나뉘게 되며, 제1형은 주로 말초신경에 종양이 많이 발생하기 때문에 말초신경형이라고도 하며, 제2형은 중추신경이나 청신경에 주로 발생하여 중추신경형이라고도 한다. 신경섬유종증 1형은 피부의 다발성 신경섬유종, 피부에 나타나는 밀크커피색 반점, 겨드랑이와 서혜부의 주근깨, 홍채의 Lisch 결절 등을 특징으로 한다[2, 3]. 이 질환의 약 50%는 NF1 유전자의 상염색체 우성에 의한 유전병으로 발생하고, 나머지 50%는 산발적으로 일어나는 돌연변이에 의해 생긴다[4]. 저자들은 국내에서 NF1 유전자의 염기서열분석에서 새로운 틀이동돌연변이(frameshift mutation)가 관찰된 2예를 보고하고자 한다.
Go to : 
증 례
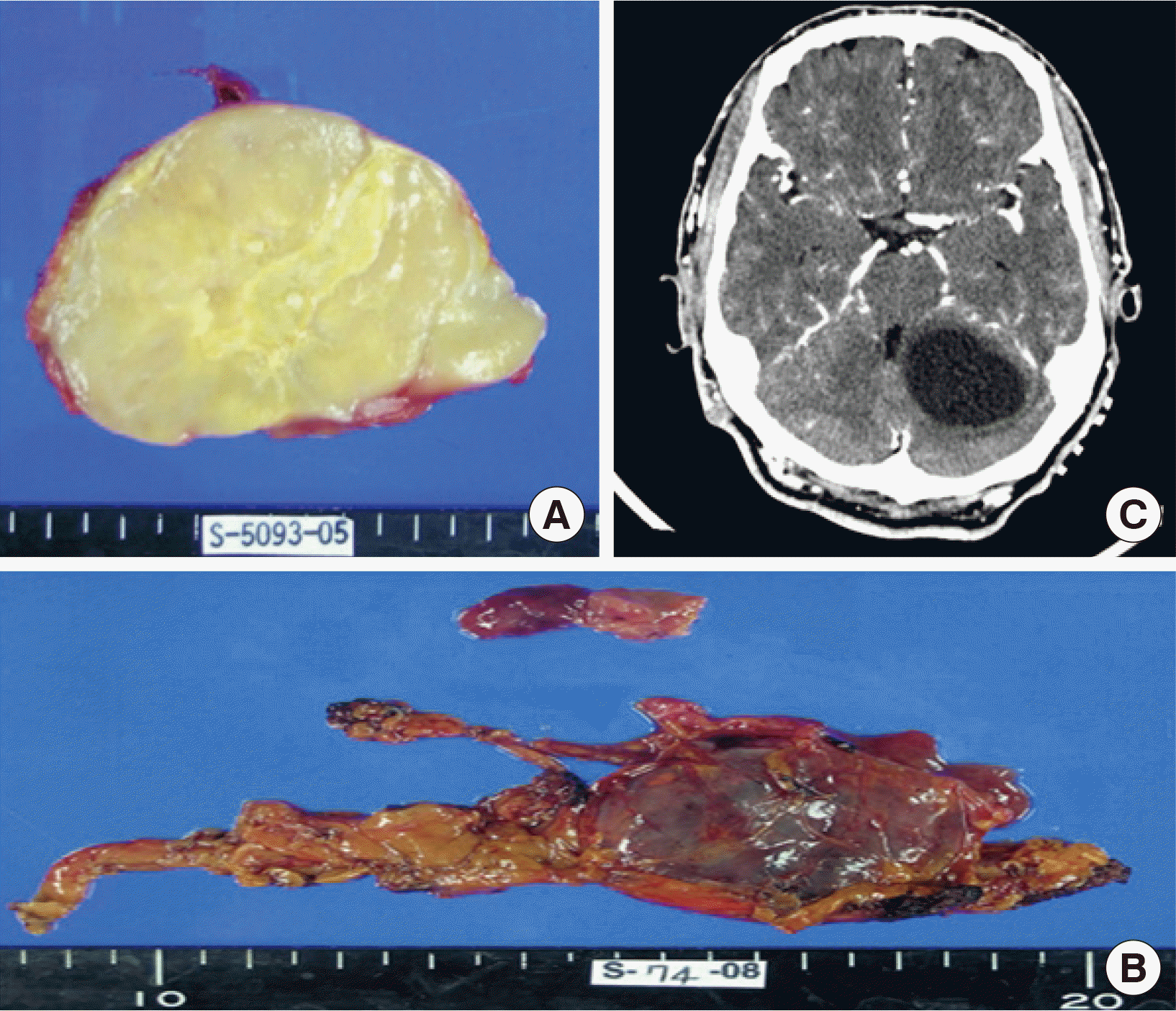
환자1은 59세 남자환자로 내원 40년 전부터 전신에 발생한 신경섬유종을 보였다. 환자 아버지가 신경섬유종증 1형이 있었다고 조사되었다. 그 외 가족에 대한 신경섬유종증 유전자 검사는 진행하지 못하였다. 환자는 내원 15년 전 오른쪽 엉덩뼈에 7 cm의 양성 종양(Fig. 1A), 내원 11년 전 오른쪽 부신에 양성종양(Fig. 1B), 내원 10년 전 좌측 소뇌에 털모양별아교세포종(pilocytic astrocytoma) (Fig. 1C)으로 각각 수술하였다. 환자는 가족력과 신경섬유종을 이유로 신경섬유종증 1형을 의심하고 있었다. DNA 추출은 DNA DSP Mini kit (QIAGEN, Hilden, Germany)을 이용하였고, 중합효소연쇄반응은 NF1 유전자 전체 엑손 58개와 인접 인트론에 대하여 PalmTaqTm High-Speed PCR Kit (Aharm Biosystem, Seoul, Korea)를 사용하였다. 중합효소연쇄반응 산물은 Biomek CleanSEQ-800rxn (Beckman Coulter, Miami, FL, USA)로 정제하였고, BigDye terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific, Waltham, MA, USA)를 이용하여 염기서열 반응을 시행하였다. 획득된 염기서열은 sequencher software v5.3 (Gene Codes, Ann Arbor, MI, USA)을 이용하여 분석하였고, 참고 염기서열을 참조하여 이상유무를 확인한 후 Human Gene Mutation Database (HGMD), Leiden Open Variation Database (LOVD)에서 변이를 확인하였다. 참고 염기서열은 NM_001042492.3과 NC_ 000017.11을 사용하였다. 병원성(pathogenic), 유사병원성(likely pathogenic), 양성(benign) 여부는 American College of Medical Genetics and Genomics (ACMG) 가이드라인을 따랐다[5]. 염기서열분석에서 단일염기다형성(single nucleotide polymorphism, SNP)인 c.702G>A, c.2034G>A가 발견되었고, 새로운 염기변이인 c.3108delA (heterozygote) (p.Lys1036Asnfs*14)이 발견되었다(Fig. 2A). ACMG 가이드라인에 따르면 이 c.3108delA는 Korean Reference Genome Database (KRGDB) 인구 집단자료에서 존재하지 않고(moderate evidence of pathogenicity, PM2), neurofibromin 기능을 상실시키는 틀이동돌연변이(very strong evidence of pathogenicity, PVS1)이며, 전신의 신경섬유종과 NF1 유전자의 연관성(supporting evidence of pathogenicity, PP4)을 보였다. 이를 종합하면 병원성(pathogenic)으로 판단할 수 있었다[5]. 이 c.3108delA는 기존에 보고되지 않은 돌연변이이다.
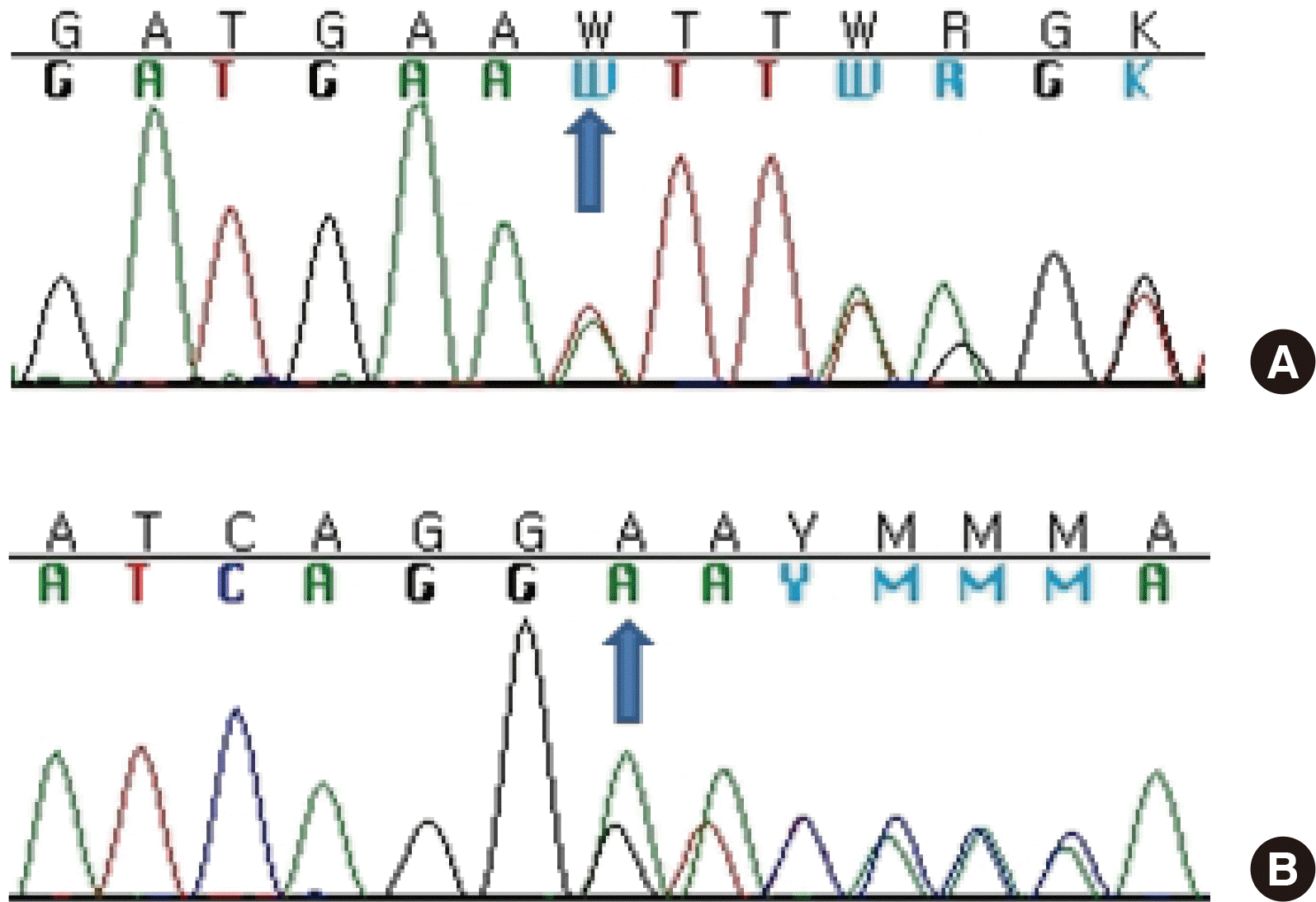
환자 2는 재태연령 38주에 체중 3 kg으로 태어났다. 환아는 25개월 여아로 출생 시부터 존재한 5 mm 이상의 밀크커피색 반점이 6개 이상 있었으며, 뇌 자기공명영상에서 특이소견은 관찰되지 않았다. 어머니 또한 밀크커피색 반점이 관찰되었으나 신경섬유종증 진단은 받지 않았다. 그 외 가족에 대한 신경섬유종증 유전자 검사는 진행하지 못하였다. 환아는 현재 갑상선기능저하증으로 하루에 levothyroxine 0.025 mg, 혈관종으로 propranolol 30 mg을 복용 중이다. NF1 유전자에서 SNP인 c.702G>A, c.2034G>A가 발견되었고, 새로운 염기변이인 c.7623delG (heterozygote) (p.Ile2541Serfs*7)가 발견되었다(Fig. 2B). ACMG 가이드라인에 따르면 c.7623delG는 KRGDB 인구집단자료에서 존재하지 않고(moderate evidence of pathogenicity, PM2), neurofibromin 기능을 상실시키는 틀이동돌연변이(very strong evidence of pathogenicity, PVS1)를 보였다. 이를 종합하면 유사병원성 이상으로 판단되었다[5]. 이 c.7623delG는 기존에 보고되지 않은 돌연변이이다.
Go to :
고 찰
신경섬유종증 1형의 가장 흔한 원인 유전자는 NF1 유전자로 17번 염색체(17q11.2)에 위치한다. 이 유전자는 DNA 크기가 350 kb 이상이며, 300 kDa의 단백질을 암호화하고 있다[6]. 이 유전자는 2,818개의 아미노산으로 이루어진 neurofibromin 단백질을 만드는 역할을 한다[2]. Neurofibromin은 말이집(myelin sheath)을 생성하는 작용을 하며, RAS proto-oncogene을 억제하는 기전으로 종양 억제 단백질의 역할을 한다. NF1 유전자에 변이가 발생하면 neurofibromin 생성이 억제되고, 이로 인해 세포분열 억제 기능이 저하되어 종양 발생이 일어나게 된다[7].
신경섬유종증 1형은 사람에서 발생하는 상염색체 우성 유전 질환 중 가장 흔하며 피부 및 신경조직에 발현한다[3]. 1988년 만들어진 진단기준에 따르면, 다음의 7가지 항목 중에서 2가지 이상을 만족하면 신경섬유종증 1형으로 진단할 수 있다[8]. 1) 6개 이상의 밀크커피색 반점(사춘기 이전에는 직경 5 mm 이상, 사춘기 이후에는 직경 15 mm 이상), 2) 2개 이상의 신경섬유종 또는 하나의 얼기상 신경섬유종, 3) 액와부와 서혜부의 주근깨, 4) 시신경교종, 5) 2개 이상의 Lisch 결절, 6) 접형골의 형성이상이나 장골의 피질이 얇아지는 것과 같은 특징적인 골병변, 7) 일차 친족에서 신경섬유종증 1형의 가족력.
신경섬유종증 1형은 개인 간, 가족 간뿐 아니라 개인의 일생에서도 시간에 따라 임상적으로 다양한 특징을 가지며, 다음에 나오는 유전자 변이와 증상만이 연관이 있는 것으로 생각된다[9]. 전체 NF1 유전자의 결손은 피부섬유종의 숫자 증가, 선천기형의 빈도증가, 체세포과다증식, 얼굴의 기형 등과 관련 있고[10-12], 17번 엑손의 3염기 결실(c.2970-2972 delAAT)은 전형적인 색소특징과 관련이 있으며[13], 29번 엑손의 암호화된 1809번째 아미노산인 arginine에 영향을 미치는 과오돌연변이(missense mutation)는 밀크커피색 반점, 학습저하, 저신장, 폐동맥 협착과 관련이 있다[14, 15]. 본 두 증례의 경우 피부병변과 가족력을 통해 제1형 신경섬유종증으로 진단할 수 있었고, 본 두 환자의 증상과 유전자변이의 직접적인 관계를 찾기는 어렵다.
신경섬유종증 1형 환자의 50% 정도는 NF1 유전자의 산발적인 돌연변이에 의해 발생하고 현재까지 3,300여개의 돌연변이가 HGMD에 보고되어 있다[3, 16]. 한국인 339가족을 대상으로 한 250개의 변이 조사에 따르면 틀이동돌연변이 114가족(33.6%), 무의미돌연변이(nonsense mutation) 98가족(28.9%), 과오돌연변이(missense mutation) 52가족(15.3%), 짜집기돌연변이(splicing mutation) 38가족(11.2%), 큰 결손(large deletion) 24가족(7.1%), 틀내 삽입/결손(inframe deletion/insertions) 13가족(3.8%)인 것으로 조사되었다[17]. 본 증례에서 발견된 c. 3108delA (p.Lys1036Asnfs*14)과 c.7623delG (p.Ile2541Serfs*7)는 틀이동돌연변이이고, 앞에서 기술한 바와 같이 틀이동돌연변이는 한국인에서 가장 흔한 돌연변이이다.
신경섬유종증 1형의 임상적 다양성은 유전적 요인, 비유전적 요인, 확률적 요인의 조합에 의해 만들어지는 것으로 보이며, 이러한 복잡성과 다양성은 유전자 이상과 표현형의 연관성 관찰을 어렵게 만들고 있어서 진단을 더욱 어렵게 한다[18]. 비록 신경섬유종증 1형은 임상적 양상만으로 진단이 가능하나, 증상이 뚜렷하지 않은 환자 혹은 소아 환자의 경우 유전자 변이의 확인을 통해 진단할 수 있다. 특히 밀크커피색 반점이나 주근깨만 있는 환자의 경우 분자진단은 Legius 증후군과 같은 유사질환의 감별을 위해 중요하다[19]. 본 증례의 두 번째 환자의 경우, 증상이 뚜렷하지 않아 분자검사가 NF1 진단에 도움이 되었다.
신경섬유종증 1형의 경우, 종양의 발생이나 척추측만증, 안과 질환 등 다양한 증상을 보일 수 있기 때문에 질환을 조기에 진단하여 환자 및 가족들의 증상과 관련된 검사를 지속적으로 시행하는 것이 중요하다. 새로운 NF1 변이의 보고는 신경섬유종증 1형 환자와 가족의 유전상담과 임상관리에 유용하게 사용될 수 있다. 이에 저자들은 신경섬유종증 1형 환자 2례에서 이전에 보고되지 않은 NF1 유전자의 돌연변이를 보고하는 바이다.
Go to :
 XML Download
XML Download