

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Fragile X syndrome (FXS; OMIM: 300624) is triggered by a large CGG repeat expansion in the 5´ untranslated region (UTR) of FMR1. The unstable large CGG expansion induces hypermethylation, which affects promoter attachment in the 5' UTR, inhibiting transcription and causing a decrease or loss of FMR1 protein [1]. FXS is the most common cause of genetic mental retardation and autism spectrum disorders, with a prevalence of approximately 1 in 5,000 males and 1 in 2,500–8,000 females [2]. Depending on the number of CGG repeats, FMR1 alleles can be categorized as full mutation (FM, >200 repeats), premutation (PM, 55–200 repeats), intermediate (INT, 45–54 repeats), or normal (NL, <45 repeats) owing to differences in the severity of symptoms and degree of maternal transmission to the next generation [3]. Therefore, diagnosis of the disease, including accurate repeat count number detection using reliable methods and allelic categorization performed according to proven classification guidelines, is very important [3-5].
Several guidelines for FXS diagnostic testing show the current difficulties in detecting all FMR1 FM characteristics using only a single method [3-5]. Conventional molecular FXS diagnostic testing consists of PCR fragment analysis (FA); Southern blotting (SB) is recommended as a complementary method. FA has the advantage of detecting the repeat number accurately, quickly, and easily. However, large CGG repeat expansions may not be amplified using PCR for GC-rich sequences. SB is considered the gold standard for FXS FM diagnosis; it can simultaneously confirm repeat expansion and methylation status using several restriction enzymes. However, it is very labor-intensive and time-consuming and is difficult to perform routinely in clinical laboratories.
Using gene-specific and CGG-specific primers, triplet repeat primed PCR (TP-PCR) can detect >200 repeats by comparison with a CGG ladder or tailing, even in large repeat expansions [6]. TP-PCR results have good concordance with conventional PCR or SB results [6, 7]. However, whether FXS can be diagnosed using solely TP-PCR has yet to be extensively studied. In the present study, using a large number of samples from Korea, the diagnostic utility of TP-PCR was compared with the conventional diagnostic protocol consisting of FA and/or SB and evaluated in terms of allele categorization, repeat number correlation, and zygosity concordance in female genetic carriers.
Go to : 
MATERIALS AND METHODS
Clinical samples and conventional diagnostic protocol
The Institutional Review Board of Seoul National University Hospital, Seoul, Korea approved this study (IRB No. 2006-194-1136). Written informed consent for the study was obtained from all participants. Blood samples were collected from a total of 458 patients (326 males, 132 females) who visited Seoul National University Hospital from November 2013 to March 2018. The collected blood samples were routinely examined for FXS, and the obtained results were analyzed retrospectively. The conventional diagnostic protocol for FXS was performed as follows: first, FA was performed; if no amplification was observed in male patients or if only one allele was observed in females, then SB analysis was performed to demonstrate higher repeat alleles in the PM or FM zone or to confirm the FA results. Additionally, each sample was analyzed by TP-PCR in parallel with the conventional diagnostic protocol to obtain CGG repeats.
FA, SB, and TP-PCR
We performed in-house FA and SA methods. In contrast, a commercial kit was used for TP-PCR developed by Asuragen. FA was performed using fluorescently labeled PCR primers F-5´-GACGGAGGCGCCGCTGCCAGG-3´ and R-5´-GTGGGCTGCGGGCGCTCGAGG-3´. The analysis was repeated with a new primer set (F-5´-CGTGACGTGGTTTCAGTGTT-3´ and R-5´-GCCGACACCAAGAAGAAAAG-3´) when the method with the former primer set showed discrepant results. Electrophoresis was performed on an ABI PRISM 3130 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) and analyzed using GeneMapper v.3.7 (Applied Biosystems) software. Experimentally, with regard to CGG repeat numbers >50, the previous FA bin set showed approximately three CGGs with lower values when measuring known CGG results without adjustment. Hence, the bin set for large expansions was adjusted based on measurement of standard materials.
For SB analysis, at least 7 μg of isolated DNA were treated with two restriction enzymes, EcoRI and EagI (NEB [New England Biolabs], Ipswich, MA, USA) and separated on a 0.7% agarose gel. The membranes were hybridized overnight with the FMR1 genomic probe StB12.3 and labeled with the Megaprime DNA Labelling System RPN 1606 (GE Healthcare Amersham, Little Chalfont, UK). After drying, the membranes were scanned using Typhoon FLA 7000 (GE Healthcare Amersham).
For TP-PCR, CGG repeat number in the 5´ UTR of FMR1 was analyzed by PCR amplification using the AmplideX PCR/CE FMR1 Kit (Asuragen, Austin, TX, USA) and agarose gel electrophoresis. Electrophoresis was performed on an ABI PRISM 3730 Genetic Analyzer (Applied Biosystems). The PCR conditions were as follows: incubating the reaction mixture at 95°C for 5 minutes, followed by 10 denaturation cycles at 97°C for 35 seconds, annealing at 62°C for 35 seconds, and extension at 68°C for 4 minutes. Next, 20 denaturation cycles at 97°C for 35 seconds, annealing at 62°C for 35 seconds, and extension at 72°C for 4 minutes 20 seconds (with 5 seconds extension at each additional cycle) were followed by final holding at 4°C. The results were analyzed using GeneMapper v.3.7 software. The repeat number was calculated using the following formula provided in the manufacturer’s instructions (Asuragen) to compensate for the migration of the GC-rich sequence:
where peaki is the size in base pairs of a given product peak; C0 is the size correction factor in ABI 3730, 50 cm configuration; and m0 is the mobility correction factor for each CGG repeats.
Statistical analysis
To analyze the repeat number correlation, we performed statistical correlation analysis using Excel and SPSS software version 21 (IBM Corp., Armonk, NY, USA). As it is impossible to obtain an exact repeat number >200 by TP-PCR, we used samples determined by TP-PCR to have repeat numbers <200 (N=361) to analyze repeat number correlation among the methods. We divided the results into two groups: 1) repeat number from 0 to 80 (N=352) and 2) repeat number from 81 to 200 (N=9) and compared them with the FA or SB results obtained from the same sample. The correlations amongst the results from different methods were accessed using Pearson’s correlation coefficient (r) and were interpreted as negligible (<0.1), weak (0.1–0.39), moderate (0.40–0.69), strong (0.70–0.89), or very strong (≥0.9) based on the absolute magnitude.
Go to :
RESULTS
Allele categorization
Of the 458 patients, allele categorization by FA and SB showed the same results as TP-PCR in FM, PM, and NL patients. Interestingly, one of the five INT patients detected by FA was classified as PM using TP-PCR. Overall, allele categorization using the two methods was highly concordant (457/458, 99.8%) (Table 1).
Table 1
Comparison of allele categorization between FA+SB and TP-PCR
| FA+SB | TP-PCR | Concordance (%) | ||||
|---|---|---|---|---|---|---|
|
|
||||||
| FM | PM | INT | NL | |||
| FA | PM (55–80) | 5 | 100 | |||
| INT (45–54) | 1† | 4 | 80 | |||
| NL (< 45) | 432 | 100 | ||||
| SB | FM (> 200) | 9 | 100 | |||
| PM (80–200) | 7 | 100 | ||||
| Total | 9 | 13* | 4 | 432 | 458 (99.8) | |
![]()
Repeat number correlation
The repeat number results determined by FA and TP-PCR showed a very strong correlation (P<0.001, r=0.987). The repeat number results determined by TP-PCR and SB showed a moderate correlation (P=0.078, r=0.614) (Fig. 1).
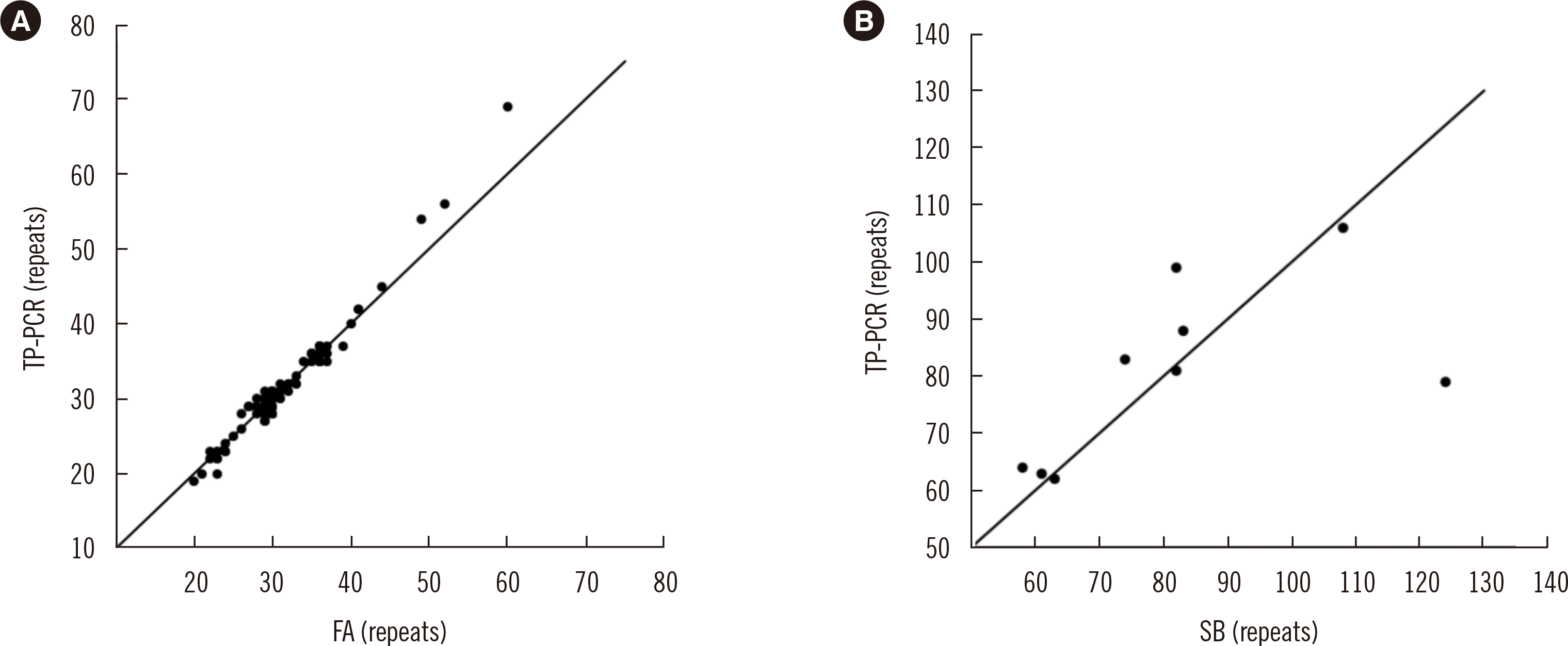 | Fig. 1Repeat number correlation based on repeat number range. The auxiliary line (y=x) in each graph is adapted for demonstrating their correlation. (A) A very strong correlation (P<0.001, r=0.987, N=352) of CGG repeat numbers was observed between FA and TP-PCR in the range from 0 to 80 of repeat numbers. (B) A moderate (P=0.078, r=0.614, N=9) correlation was observed between SB and TP-PCR in the range from 81 to 200 of repeat numbers., as obtaining an accurate CGG repeat value using SB is difficult compared with TP-PCR.
Abbreviations: FA, fragment analysis; SB, Southern blotting; TP-PCR, triplet repeat primed PCR.
|
Zygosity concordance in female genetic carriers
TP-PCR evaluation to discriminate each allele separately in females (N=132) revealed high concordance (88.3%) and 22 discordant results (12.7%) between the methods (Table 2). Of the 22 discordant cases, 10 were detected as homozygote by FA but as heterozygote by TP-PCR, and the remaining (N=12) were detected as homozygote by TP-PCR but as heterozygote by FA. In eight out of 10 cases detected as homozygote by FA and heterozygote by TP-PCR, TP-PCR detected one additional PM or FM allele that was not detected by FA under normal PCR conditions. These eight cases were further analyzed using SB to confirm the presence of the additional alleles detected by TP-PCR. The TP-PCR and SB results were concordant in all cases. Therefore, these results further confirmed that TP-PCR has greater sensitivity than FA for detecting zygosity. In contrast to these eight cases, two cases showed two heterozygous normal alleles (NLs) by TP-PCR, but only one NL by FA (Table 2).
Table 2
Comparison of zygosity between FA and TP-PCR
| TP-PCR | Total | |||
|---|---|---|---|---|
|
|
||||
| Heterozygote | Homozygote | |||
| FA | Heterozygote | 76 | 12* | 88 |
| Homozygote | 10† | 34 | 44 | |
| Total | 86 | 46 | 132 | |
*All 12 cases had two alleles corresponding to adjacent peaks (28/29 or 29/30); TP-PCR redone using a 10-fold dilution clearly showed distinct two peaks. †Of the 10 cases, eight had one PM or FM allele, as well as one NL, which was missed by FA. The other two cases were heterozygous NLs detected using TP-PCR, whereas only one NL was detected using FA. Abbreviations: FA, fragment analysis; TP-PCR, triplet repeat primed PCR; NL, normal allele; PM, premutation (allele); FM, full mutation (allele).
![]()
In the latter 12 cases, the peaks were wider and blunt or split/forked compared with those observed in the regular cases. In FA, the two peaks were located close to each other, with a gap of one repeat number (29/30, mostly). We hypothesized that that relatively high concentration of DNA (30 μg/mL) used to perform capillary electrophoresis during TP-PCR compared with FA (approximately 10 μg/mL, used after five-fold dilution of an original concentration of 50 μg/mL) might have affected the shapes of the two adjacent peaks in TP-PCR, causing them to appear as one merged peak. Accordingly, these inconsistent cases were analyzed again by TP-PCR with a 10-fold dilution; the results clearly identified these cases as heterozygotes, consistent with the FA results (Fig. 2).
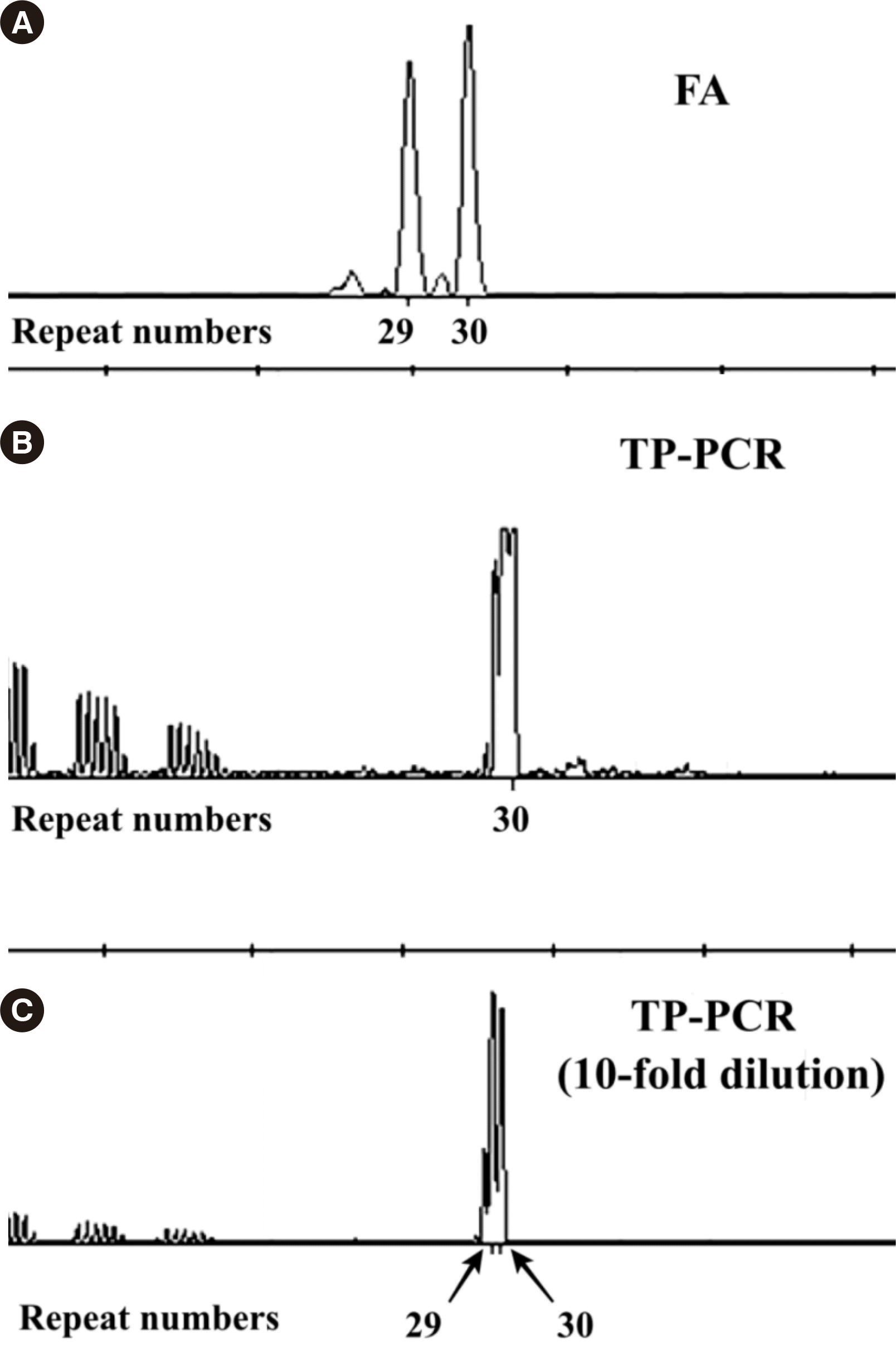 | Fig. 2Zygosity discrepancy between FA and TP-PCR and peak separation using a diluted DNA sample for TP-PCR. (A, B) One patient had 29 or 30 repeats when using FA but only 30 repeats using TP-PCR. (C) Allele separation was achieved, and two separate peaks were obtained using TP-PCR with a 10-fold dilution of the DNA sample.
Abbreviations: FA, fragment analysis; TP-PCR, triplet repeat primed PCR.
|
Mosaicism
As shown in Table 3, of the five size mosaicism cases, no amplification was observed for three males (numbers 1–3) by FA; however, the TP-PCR results showed two or three peaks in the PM/FM range. Interestingly, the last case (number 5) demonstrated a complex mosaicism pattern of NL, PM, and FM alleles by TP-PCR. However, the band indicating an FM allele was not observed by SB; instead, a smeared, continuous band spectrum was observed (Fig. 3), which has been observed for unmethylated FM alleles [8]. This result demonstrates the concordance between the TP-PCR and SB results. Thus, we could diagnose this case as size mosaicism in a female carrier with three alleles: NL, PM, and an unmethylated FM allele. Compared with FA, TP-PCR could detect hidden PM and FM alleles, which are not amplified by FA, and the TP-PCR results also showed perfect concordance with the SB results (Fig. 3).
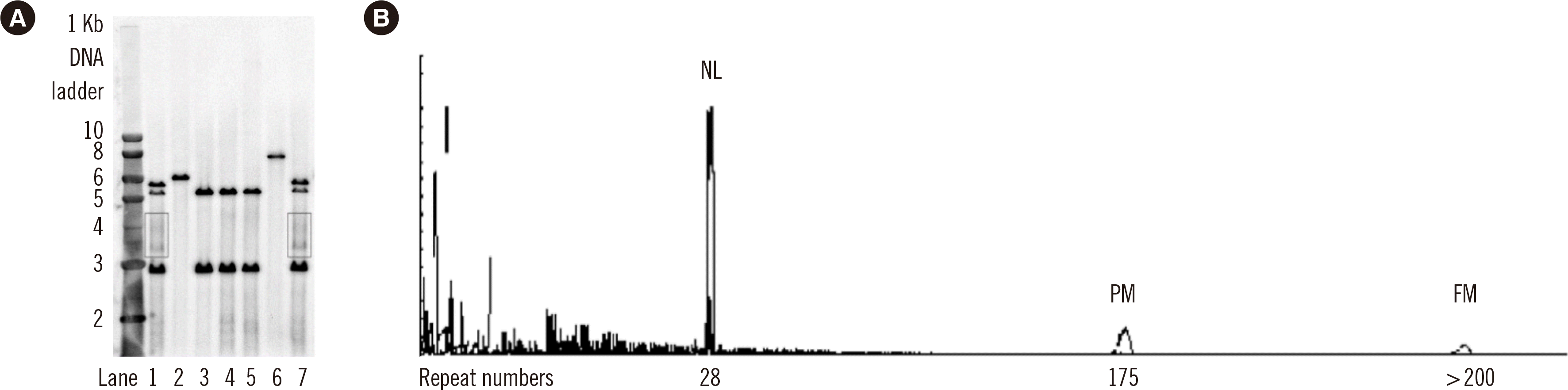 | Fig. 3Mosaicism cases in fragile X syndrome. (A) SB results. Lanes 1 and 7 are the results of patient P4 and show mosaicism with NL, PM, and unmethylated FM. Smeared, continuous bands are observed in the boxed areas, indicating an unmethylated FM allele. Lanes 2 and 6 show FM in male carriers; Lanes 3, 4, and 5 show NL in females. (B) TP-PCR result of patient P4, with three very clear peaks in the areas of NL, PM, and FM.
Abbreviations: SB, Southern blotting; TP-PCR, triplet repeat primed PCR; NL, normal allele; PM, premutation (allele); FM, full mutation (allele).
|
Table 3
Mosaicism cases in fragile X syndrome
| Case No. | Sex | FA | SB | TP-PCR |
|---|---|---|---|---|
| 1 | M | Not amplified | 156, 436, 951 | 156 and two other peaks in > 200 |
| 2 | M | Not amplified | 120, 623 | 120 and two other peaks in > 200 |
| 3 | M | Not amplified | 351, 563, 897 | three peaks in > 200 |
| 4 | F | 30 | 30, 83, 93 | 30, 88, 93 |
| 5 | F | 29 | 30, 160, 204* | 28, 175 and another peak in > 200 |
![]()
Go to :
DISCUSSION
We examined whether TP-PCR has better accuracy than conventional methods in detecting FXS. We compared its performance with that of FA and SB in terms of allele categorization, repeat number correlation, and zygosity determination in females. TP-PCR showed superior results compared with the conventional methods, indicating that it is reliable for FXS diagnosis.
Discrepancies in allele categorization were observed in one patient; GC-rich DNA, such as CGG repeat expansion in FXS, is known to migrate faster than the size standard in capillary electrophoresis [9, 10]. To adjust for this phenomenon, bin set adjustment was performed using predetermined exact size control material. In cases of CGG repeat number >50, FA underestimated the values compared with TP-PCR. All allele categorization results were consistent, except for one case, in which a CGG repeat number of 54 and 55 (on the border of the INT and PM diagnostic criteria) was determined by FA and TP-PCR, respectively. Additionally, the recommendations of the College of American Pathologists/American College of Medical Genetics Biochemical and Molecular Genetics Committee for FXS testing clearly demonstrated the technical limitations of size analysis, with an acceptable range of ±5 (<55 repeats) or ±10 (56–100 repeats) as the consensus size [3]. Establishing laboratory guidelines is necessary to overcome the limitations of this method. For example, one approach involves additional assessment for AGG interruption. As reported in several studies, lack of AGG interruption is an important mechanism of FXS CGG expansion and its transmission to offspring [11, 12]. Normal AGG interruption (3–4) is anchored after every 10–12 CGG repeats. The procedural loss of AGG interruption is thought to induce slippage of CGG expansion during meiosis. Thus, PM or higher repeat number INT carriers usually show loss of AGG interruption (0–1). Using this approach, the risk of transmission to the next generation can be more accurately determined. Physicians can apply AGG information to predict the risk in patients with mildly expanded alleles, which could be helpful for INT and PM carriers interested in prenatal or preimplantation genetic diagnosis.
With respect to repeat number correlation, our results demonstrated that TP-PCR can be used to determine the exact repeat number in cases of <200 repeats. Very high correlation was observed when the TP-PCR and FA results were compared for low repeat numbers. In cases of repeat numbers approximately >50 CGG, FA underestimated the values compared with TP-PCR, although FA bin size adjustment was performed. These discrepancies are most likely due to the capillary migration speed in FA. In addition, TP-PCR was shown to accurately detect PM and FM compared with the SB results for relatively higher repeats. Therefore, TP-PCR can adequately replace established methods for all CGG repeat ranges.
Determining accurate zygosity is very important in female genetic carriers. FXS, unlike other Mendelian disorders, reportedly demonstrates fragile X-associated primary ovarian insufficiency in female PM carriers, despite the absence of the FM allele [13]. In the present study, we repeated the analysis to confirm whether the extra alleles observed by TP-PCR were an artifact; however, the results were the same as the previously obtained ones. In addition, we checked for allele dropout using a new primer set, considering the possibility of variants or deletions in the current FA primer binding sites. However, only one normal allele per case was observed. Although we could not determine why FA failed to detect the other alleles, we hypothesize that TP-PCR can detect alleles much more sensitively, because the DNA concentration used in TP-PCR is higher than that used for FA. In addition, if these two cases have small population sizes of mosaicism, it can be detected by TP-PCR but not by FA. These results are similar to previously reported cases demonstrating different results for FA and TP-PCR, which were attributed to different DNA amounts [14].
To clarify discrepancies between FA and TP-PCR, we first repeated the analysis with a diluted sample and succeeded in separating the single peak into two heterozygous peaks. Scanning the electropherogram in advance, before determining whether peak shapes are atypical, could aid in the general laboratory process; the required samples could be diluted to confirm heterozygosity.
In addition, TP-PCR showed good detection ability for size mosaicism. TP-PCR identified the FM alleles and accurately detected those observed in the SB results. In case number 4, the smear band spectrum that could not be distinguished as a band owing to blurring in SB gel electrophoresis was determined as an FM allele based on the TP-PCR results.
TP-PCR has a few limitations. First, although TP-PCR can detect the presence of FM, it is difficult to confirm an exact repeat number ≥200. However, unlike PM, the severity of symptoms in FM patients has not been reported to increase with increasing repeat number [15]. In a study examining the relationship between repeat number and intelligence in a woman with only FM alleles, length of expansion and intelligence were not associated [16]. Second, methylation cannot be confirmed using TP-PCR. The FM allele is most highly associated with hypermethylation within CpG islands, leading to FMR protein (FMRP) depletion. However, in a proportion of males carrying the FM allele, the CpG islands are only partially methylated; these males are less affected than FM males with fully methylated CpG islands. In addition, several patients have a complete lack of methylation but a CGG repeat number >200. Patients with a lack of methylation typically have a higher level of FMRP and are often high functioning with an intelligence quotient within or bordering the normal range [17, 18]. In most cases, hypermethylation in FM is typical; however, if the patient is suspected as having FXS but shows a milder phenotype or nearly normal characteristics by TP-PCR, the methylation status needs to be confirmed using SB or methylation-specific TP-PCR.
In summary, we demonstrated that TP-PCR shows superior performance compared with conventional methods. TP-PCR could serve as a sole screening method of choice for FA or FA and/or SB and that it might be reasonable to perform SB or other methods to determine methylation status only when FM alleles are detected.
Go to :
 XML Download
XML Download