

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Microchimerism including donor-derived cell-free DNA (ddcfDNA) can be used for predicting organ rejection after transplantation [1]. For the analysis of transplant rejection, it is necessary to distinguish between the donor and the recipient, hence, there is need to select a marker from single nucleotide polymorphisms (SNPs), short tandem repeats (STRs), or human leukocyte antigen (HLA) testing [2]. STRs are highly polymorphic and are used as markers to distinguish between individuals; however, disadvantages of STR analysis include high cost as it involves sequencing and longer duration than polymerase chain reaction (PCR) analysis [3]. SNPs are bi-allelic and can be used as a combination of several useful SNP markers; they have an advantage of reduced cost compared to sequencing methods as PCR is used for SNP analysis. The development of the highly sensitive droplet digital PCR (ddPCR) method enables cheaper and faster analysis compared to sequencing [2]. ddcfDNA can be detected by ddPCR using a SNP marker to predict early rejection after transplantation [4].
Serial inspection using markers of acute injury must be performed for early detection of rejection after solid organ transplantation. Therefore, it is practical to use an inexpensive PCR-based method with performance similar to the STR analysis method. This is a study to select markers to differentiate donor and recipient to measure ddcfDNA and recipient-derived cell free DNA. In order to compare the usefulness of SNPs to STR, we selected SNPs that enabled us to distinguish donor-recipient and compared them with commercially available STR.
METHODS
The study’s protocol was reviewed and approved by the Institutional Review Board of Pusan National University Yangsan Hospital (IRB No. 04-2019-006) and written informed consent was obtained from all participants.
Patients
We tested DNA of patients from January 2016 to May 2019, comprising 52 donors and 52 recipients of solid organ transplantation, with organ transplants including liver, kidney, heart, lung, and pancreas. SNP markers were selected to differentiate donor DNA from recipient DNA. The study was conducted in compliance with the principles of the Declaration of Helsinki.
Genomic DNA Extraction
Whole blood samples were collected from the donors and recipients. Extraction of genomic DNA from peripheral blood was performed using the QIAamp DNA blood mini kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions.
SNP Assays
All selected SNPs complied with the following criterion: known and validated minor allele frequency (MAF) ≥43% in Korean and all reported ethnicities (data from public databases: HapMap, 1000 Genomes, and the Korean Reference Genome project). Assuming Hardy-Weinberg equilibrium, a SNP with a MAF between 0.4 and 0.5 would be homozygous for each allele in both donor and recipient about 22% to 25% of the time. The probability of both donor and recipient having a different allele is therefore 11.5% to 12.5%, using accepted estimation models for calculation of exclusion probabilities [5]. A set of 22 assays targeting different SNPs were performed for 52 donor and recipient pairs. A TaqMan probe labeled with fluorescein amidite at the 5′-end nucleotide, and with a quencher (Black Hole Quenchers; Sigma-Aldrich, St. Louis, MO, USA) at the 3′-end nucleotide was used. SNP genotyping was performed using the QuantStudio real-time PCR (qPCR) instrument. The 20 μL PCR mixture contained Taqman Genotyping Master Mix (10 μL), Taqman Genotyping Assay (20X, 1 μL), DNase-free, RNase-free water (7.5 μL), and genomic DNA (1.5 μL). The reaction conditions were as follows: (95°C for 10 minutes)/(95°C for 15 seconds and 60°C for 1 minute) 40 cycles.
STR Assays
STR assays were performed using the AmpFLSTR Identifiler Plus PCR Amplification Kit (Applied Biosystem, Foster City, CA, USA) consisting 16 tetranucleotide repeat loci (D3S1358, vWA, D8S1179, D21S11, D18S51, D5S818, D13S317, D7S820, CSF1PO, TH01, D16S539, D2S1338, D19S433, TPOX, and FGA) and the Amelogenin gender-determining marker (AMEL). Each PCR reaction was performed with a final volume of 25 μL containing 10 μL Identifiler Plus Master mix, 5 μL Identifiler Plus Primer Set, and 10 μL genomic DNA (10 μL, 0.1 ng/μL) or control (10 μL, 0.1 ng/μL). The PCR reaction conditions were as follows: 11 minutes at 95°C; followed by 20 seconds at 94°C, 3 minutes at 59°C, and 10 minutes at 60°C for 29 cycles; and a final hold at 4°C. A 10 μL sample contained 8.7 μL Hi-Di Formamide, 0.3 μL gene scan 500 LIZ Size Standard, and 1 μL of PCR product or an allelic ladder denatured at 95°C for 3 minutes. PCR products were analyzed on an ABI Prism 3500 DNA sequencer (Applied Biosystems, Foster City, CA, USA) using the GeneMapper software 5 (Thermo Fisher Scientific, Waltham, MA, USA). The PCR products, which were 280–312 bp in size were analyzed with the GeneScan software.
Analysis of SNP Marker Performance
We counted the number of markers showing informative genotypes. The SNP haplotypes of a minor DNA contributor that are nonshared with the major DNA represents the DNA profile that can be detected in the mixture of donor and recipient DNAs. Based on Hardy-Weinberg assumptions, the probability of informative genotypes (I) at a given SNP marker can be calculated as I=2a2b2+2a3b+2ab3, where a and b are the frequencies of the A and B alleles, 2a2b2 is the probability that the major and minor DNA contributors are homozygous for the opposite allele (a2b2+a2b2), and 2a3b+2ab3 are the probabilities that the major DNA contributor is homozygous (either A or B) and the minor DNA contributor is SNP heterozygous, respectively ([a2[2ab]+b2[2ab]). The I value for the SNP markers is reported in Table 1. If the I value is 0.5, there is a 50% chance of distinguishing the donor/recipient at the type I level of that SNP. The higher the I of the SNP, the less the number of SNPs required, and the donor/recipient can be distinguished only with the type I marker.
Analysis of STR
The probability of identity (Pi), also referred to as the matching probability, is the chance that two unrelated people selected at random will have the same genotype and was first described by George Sensabaugh in 1982. The Pi value of a single locus is determined by summing the square of the observed genotype frequencies:
where Xi is the genotype frequency.
Lower Pi values indicate more variability of the genetic marker in the measured population because there are more genotypes occurring at a lower frequency.
Informative Maker Analysis
The SNP or STR loci of informative markers were analyzed. Type I markers, or loci type I, were defined as loci or alleles presenting a homozygous genotype in the recipient and an alternative homozygous genotype in the donor. Conversely, type II markers were defined as loci or alleles presenting a homozygous genotype in the recipient and a heterozygous in the donor. Type III markers were those that did not fulfil the criteria for type I or type II markers.
RESULTS
DNA from 52 donor recipient pairs were tested for informative markers. The number of deceased donors was 28 and the number of living donors was 24. Males (n=30, 57.7%) outnumbered females in the total number of recipients (Table 2) and the median age of recipients was 47 years. The MAF of SNPs in the 52 pairs were 1.0%–76.0% (Table 1). The probability of informative genotypes (I) ranged from 0.001–0.124 and the summation of I was 0.680. In all donor recipient pairs, the selected SNP markers had an average probability of being informative of 0.031. After the exclusion of the least polymorphic markers (rs1265094, rs10228737, rs7072759, rs1467245, rs10164176), the average probability of being informative increased to 0.039.
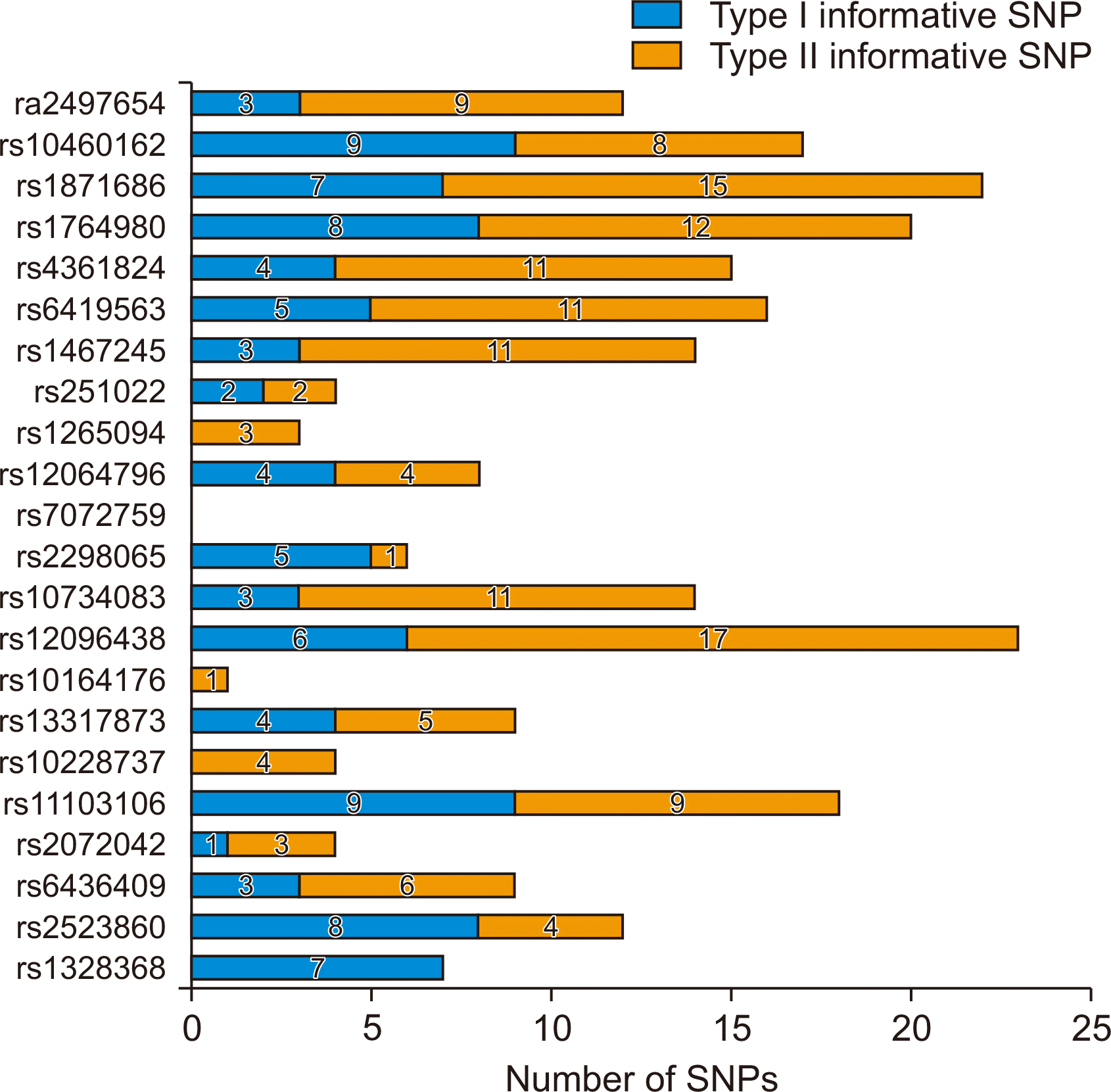
The most selected type I informative markers were rs10460162 and rs11103106, observed in 9 donor recipient pairs (17.3%), followed by rs2523860. Four SNPs, rs10228737, rs10164176, rs7072759, and rs1265094, had no discriminatory power as type I informative markers. The most selected type II informative marker was rs12096438 observed in 17 donor recipient pairs (32.7%) (Fig. 1).
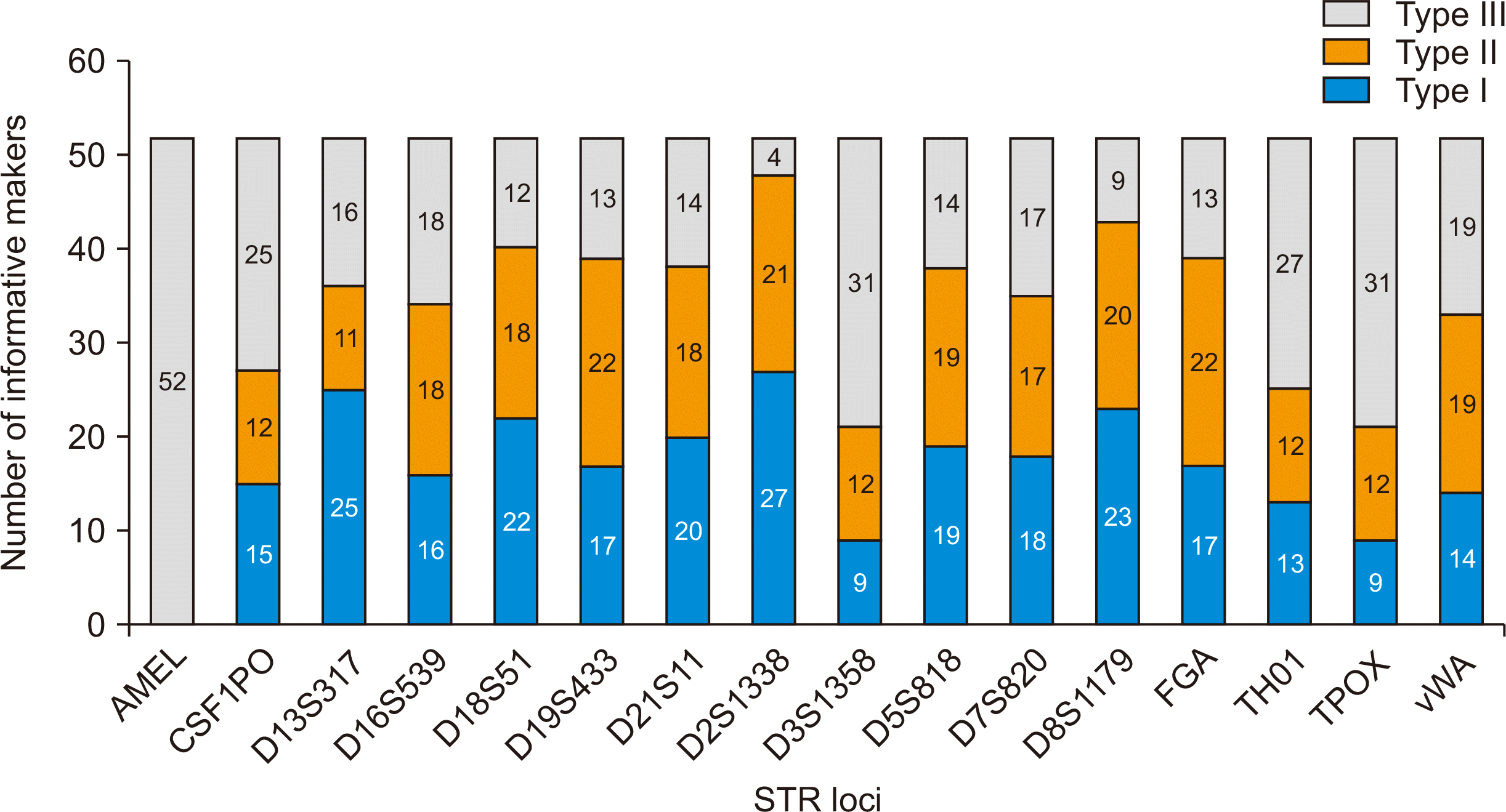
The Pi or matching probability is the chance of the same genotype in two unrelated people selected at random. The Pi of the present study ranged from 0.122 to 0.348 (Table 3). AMEL was not an optimal marker for the determination of mixed chimeras due to the sharing of alleles between donor and recipient. Except AMEL, the 15 STR loci were informative markers with a discrimination power of 42.9%–92.9%. The selection rate of type I informative markers among 16 alleles was 17.1% to 56.1%, with D2S1338 observed in 27 donor recipient pairs (Fig. 2). There were no type I alleles in two mother-child pairs, one sister-sister pair, and one uncle-nephew pair.
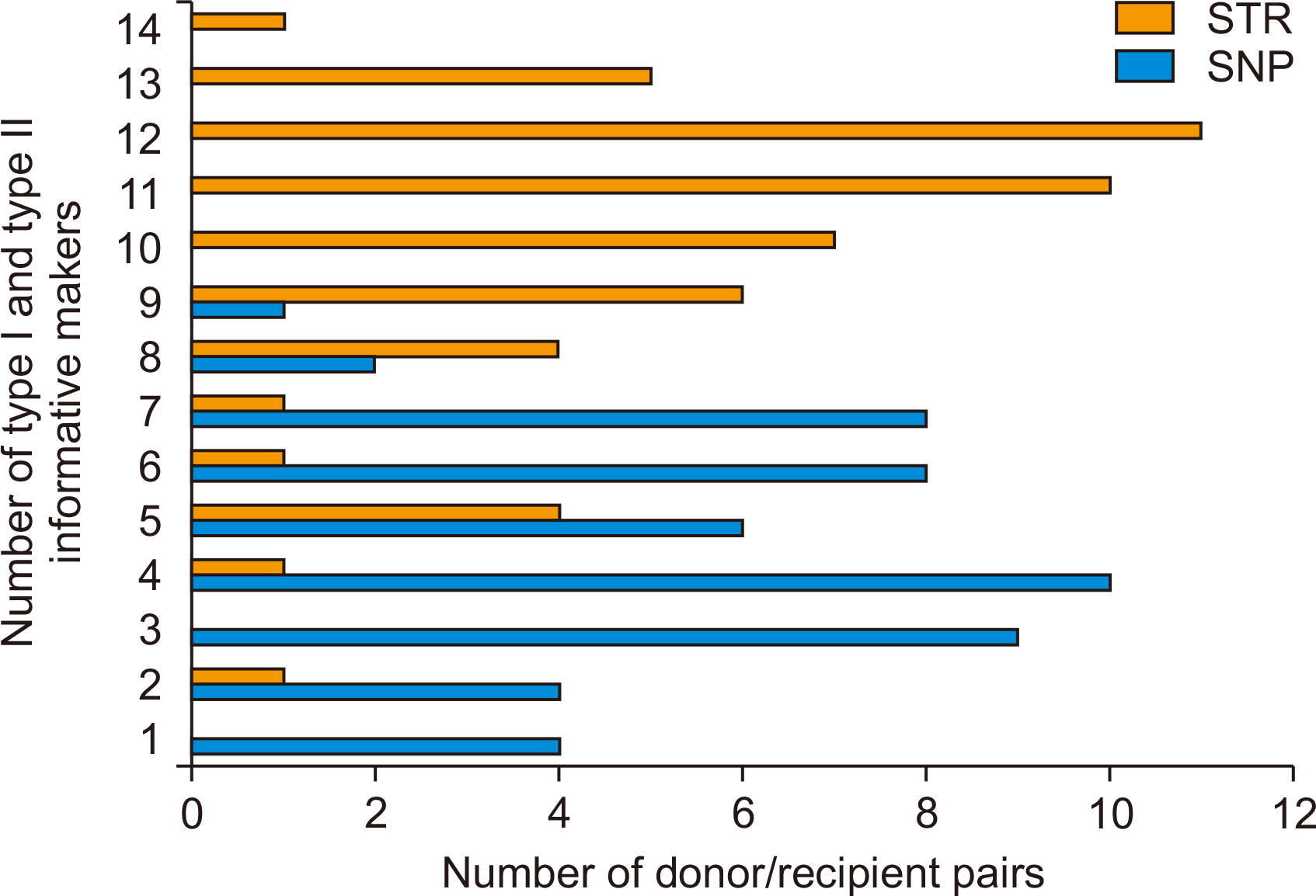
In 52 pairs of donors and recipients, the median values of type I SNPs were 2 and the median values of type II SNPs were 3. The median value of type I STR was 6 and type II STR was 5. The sum of meaningful markers on combining type I and type II was median 4 (range, 1–9) in SNP and median 11 (range, 2–15) in STR (Fig. 3).
DISCUSSION
The development of rejection after transplantation is a very important event leading to loss of organ transplant, and hence, early detection of rejection is important for organ transplant survival. The use of noninvasive and sensitive test methods for diagnosis of rejection have shown that an increase in amount of ddcfDNA beyond a certain level after organ transplantation can be used as a predictor of transplant rejection [5,6]. This is a preliminary study for a follow-up study to predict organ transplant rejection by measuring ddcfDNA in recipients after organ transplantation. Variation among races should be considered when using the SNP method [7,8]. In order to compare the usefulness of SNPs as markers, we selected SNPs to distinguish between donors and recipients in a Korean population by comparing them with STR markers that are commercially available. Of the 22 SNPs, 17 markers could be configured as the primary marker set, and the excluded SNPs included 5 SNPs. Four SNPs, rs10228737, rs10164176, rs7072759, and rs1265094, in which donor/recipient pairs could not be differentiated, as well as rs1467245, with an I value of 0.005 or less were excluded. All donor ddcfDNA were categorized in STR as well as SNP and were classified into 52 pairs (100.0%).
Several tests are widely used in forensic science to distinguish between individuals. HLA present on chromosome 6 has a wide variety and is inherited as a haplotype [9]. The probability of HLA agreement is high in kidney transplantation, and hence is not suitable for discrimination between donor and recipient post transplantation. STRs are the first choice of genetic markers in forensic science as their use yields a satisfactory result in almost all cases, mainly due to their high degree of polymorphism and consequent discrimination power [3]. STRs are multiallelic, and the probability of two test objects coinciding is almost zero if many markers are used. STRs vary depending on the marker composition but the probability of a match or the likelihood of a match is approximately 0.0001 to 0.000005 or 1012 power [8]. The probability of STR match is very low, and hence the large ability to distinguish between two individuals. In our study, the probability of difference between donor and beneficiary using the STR set of 16 markers was 2.6478E-12.
SNPs are genetic markers that consist DNA sequence variations resulting from single base changes in the genome sequence, and have been considered as additional informative markers to provide reliable likelihood ratios (LRs) [3]. Most SNPs are bi‐allelic and exhibit a maximum heterozygosity of 0.50. The most efficient marker of the ideal heterozygosity of 0.50 has 37.5% probability of distinguishing two test subjects [10]. However, even though markers with an MAF between 0.4 and 0.5 are selected, the probability of one marker varies depending on the number of evaluations (N), so the set configuration differs among the analyzers. On an average, our markers show 0.039 probability of being informative, after the exclusion of the least polymorphic marker. In the present study, a MAF of 0.4–0.5 in the global population and a MAF of 0.4–0.5 in the Korean population were studied. However, the MAF of the study subjects was different from the documented global and Korean MAFs. In addition, some Korean markers had a MAF less than 0.4, resulting in lower I value.
There is need of a sensitive assay as the amount of ddcfDNA is small compared to the amount of recipient DNA. This can be expressed as a detection limit, i.e., if the detection limit is 1 ng, it means that substances present below 1 ng cannot be detected. Therefore, a test method with a low detection limit is essential for ddcfDNA detection. The detection limit for the test methods is 0.1–1 pg for qPCR, 1 fg for digital PCR, and 0.05 to 0.1 ng for STR. Digital PCR can detect the smallest amount of DNA [11,12]. In digital PCR, the sample is divided into many independent partitions. The distribution of target sequences in the partitions can be approximated with a Poisson’s distribution. Each partition acts as an individual PCR micro reactor and the partitions containing amplified target sequences are detected by fluorescence. The ratio of positive partitions over the total number allows determination of the concentrations of the target DNA in the sample [13]. Due to this principle, the sensitivity of digital PCR is 0.001% [14]. Development of digital PCR has allowed a low detection limit and increased sensitivity, hence studies are underway to detect ddcfDNA for monitoring rejection after transplantation.
The study uses the PCR method, which is cheaper than the STR method (sequencing method), is easy to test, with a shorter test time [7]. STR can be quantitatively measured and is used to measure mixed chimerism in stem cell transplantation patients [15]. Sensitivity is important to detect mixed chimerism; thus, it is advantageous to use ddPCR, since it can detect a very small amount of ddcfDNA, and the detection amount can be quantitatively analyzed [16,17].
An obvious limitation of the current study is the small size as only 52 donor/recipient pairs were enrolled. Therefore, SNP configuration requires further validation by discovering additional markers and expanding SNP research in the Korean population.
In our study, donors and recipients were distinguished by STR and 22 SNPs with MAF identified from SNP databases. With the exception of five SNPs, 17 SNP markers were able to distinguish both donors and recipients. This finding could be further used to predict organ damage by identifying ddcfDNA in the recipient's blood after solid organ transplantation.
 XML Download
XML Download